COVID-19 Guideline, Part 1: Treatment and Management
COVID-19 Guideline, Part 2: Infection Prevention
COVID-19 Guideline, Part 4: Serologic Testing
COVID-19 Guideline, Part 5: Antigen Testing
Management of Drug Interactions With Nirmatrelvir/Ritonavir (Paxlovid®): Resource for Clinicians
Mary K. Hayden,* Kimberly E. Hanson, Janet A. Englund, Mark J. Lee, Mark Loeb, Francesca Lee, Daniel J Morgan, Robin Patel, Ibrahim K El Mikati, Shahad Iqneibi,** Farouk Alabed,** Justin Z. Amarin,** Razan Mansour,** Payal Patel,** Yngve Falck-Ytter,** Rebecca L. Morgan,** M. Hassan Murad,** Shahnaz Sultan,** Adarsh Bhimraj, Reem A. Mustafa**
*Corresponding Author **Methodologist
September 5, 2023
Version 3.0 has been released and contains recommendations for SARS-CoV-2 nucleic acid testing based on new systematic reviews of the diagnostic literature.
This update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the American Society for Microbiology.
Update History
December 23, 2020
Version 2.0.0 of the guideline has been released.
May 6, 2020
Version 1.0.1 of the guideline has been released.
Abstract
Background: Accurate molecular diagnostic tests are necessary for confirming a diagnosis of coronavirus disease 2019 (COVID-19). Direct detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) nucleic acids in respiratory tract specimens informs patient, healthcare institution and public health level decision-making. The numbers of available SARS-CoV-2 nucleic acid detection tests continues to increase rapidly, as does the COVID-19 diagnostic literature. Thus, the Infectious Diseases Society of America (IDSA) recognized a need for frequently updated systematic reviews of the literature to inform evidence-based best practice guidance.
Objective: The IDSA’s goal was to develop an evidence-based diagnostic guideline to assist clinicians, clinical laboratorians, patients and policymakers in decisions related to the optimal use of SARS-CoV-2 nucleic acid amplification tests. In addition, we provide a conceptual framework for understanding molecular diagnostic test performance, discuss nuances of test result interpretation in a variety of practice settings and highlight important unmet research needs related to COVID-19 diagnostic testing.
Methods: IDSA convened a multidisciplinary panel of infectious diseases clinicians, clinical microbiologists, and experts in systematic literature review to identify and prioritize clinical questions and outcomes related to the use of SARS-CoV-2 molecular diagnostics. Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology was used to assess the certainty of evidence and make testing recommendations.
Results: The panel agreed on 12 diagnostic recommendations.
Conclusions: Access to accurate SARS-CoV-2 nucleic acid testing is critical for patient care, hospital infection prevention and the public response to the COVID-19 pandemic. Information on the clinical performance of available tests continues to grow, but the quality of evidence of the current literature to support this updated molecular diagnostic guideline remains moderate to very low. Recognizing these limitations, the IDSA panel weighed available diagnostic evidence and recommends nucleic acid testing for all symptomatic individuals suspected of having COVID-19. In addition, testing is suggested for asymptomatic individuals with known or suspected contact with a COVID-19 case when the results will impact isolation/quarantine/personal protective equipment (PPE) usage decisions. Evidence in support of rapid testing and testing of upper respiratory specimens other than nasopharyngeal (NP) swabs, which offer logistical advantages, is sufficient to warrant conditional recommendations in favor of these approaches.
Keywords: coronavirus, SARS-CoV-2, COVID, COVID-19, pneumonia, diagnosis, diagnostics, RNA testing, specimen
Executive Summary and Background
Executive Summary
Molecular diagnostic testing, i.e., nucleic acid amplification testing (NAAT)) has played a critical role in the global response to the COVID-19 pandemic. Accurate SARS-CoV-2 NAATs are needed to inform patient management decisions, hospital infection prevention practices, and public health responses. Detection and quantification of SARS-CoV-2 RNA over the course of infection has also been essential for understanding biology of disease. Given the rapid expansion of the COVID-19 molecular diagnostic literature along with increasing test availability, the IDSA recognized the need for frequently updated, evidence-based guidelines to support clinicians, clinical microbiologists, patients and policy makers in decisions related to the use of SARS-CoV-2 diagnostics.
In this second update, the IDSA panel focused on clinically relevant questions for which new data might be available to inform a new recommendation or to change the direction or strength of an earlier recommendation. Recommendations related to testing specimens from the upper versus lower respiratory tract in patients with lower respiratory tract disease, and recommendations related to testing of immunocompromised individuals or prior to initiation of immunocompromising therapies were retired. New questions were added that asked whether molecular testing was useful in guiding timing of release from isolation, or eligibility for surgical or medical procedures, in persons with a recent history of SARS-CoV-2 infection (Recommendations 10 and 11). The IDSA panel also included a new question about the accuracy of repeat testing in asymptomatic persons, and a question about home testing. Regarding reassessment of earlier recommendations, although the certainty of evidence increased for most recommendations, only one changed direction. In the earlier update, RNA testing for asymptomatic individuals who were planning to undergo surgery was suggested, whereas in the current update routine testing of these individuals is suggested against (Recommendation 9 below). For most recommendations, data were limited or absent on the clinical or analytical performance of SARS-CoV-2 NAATs in immunocompromised or vaccinated individuals, in those with a prior SARS-CoV-2 infection, in children, and in patients infected with newer SARS-CoV-2 variants, e.g., Omicron. In total, the IDSA panel made 12 recommendations for SARS-CoV-2 nucleic acid testing based on new systematic reviews of the diagnostic literature. An updated algorithm based on these recommendations is provided to aid in decision-making (see Figure 1). Recommendations assumed availability of diagnostic tests and specimen collection devices. Based on reviews of baseline risk, assumptions were made about COVID-19 disease prevalence in the community and/or pretest probabilities in individual patients, both of which influenced testing recommendations.
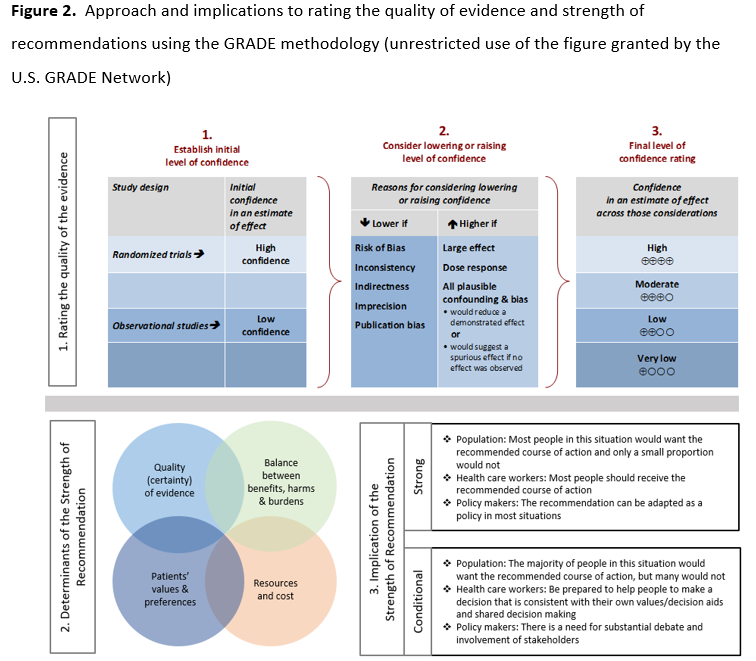
A detailed description of background, methods, evidence summary and rationale that support each recommendation, and research needs can be found online in the full text. Briefly, an expert panel consisting of clinicians, medical microbiologists, and methodologists critically appraised the COVID-19 diagnostic literature using Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology to assess the certainty of evidence. Per GRADE, recommendations are categorized as “strong” or “conditional.” The word “recommend” indicates strong recommendations and “suggest” implies conditional recommendations.
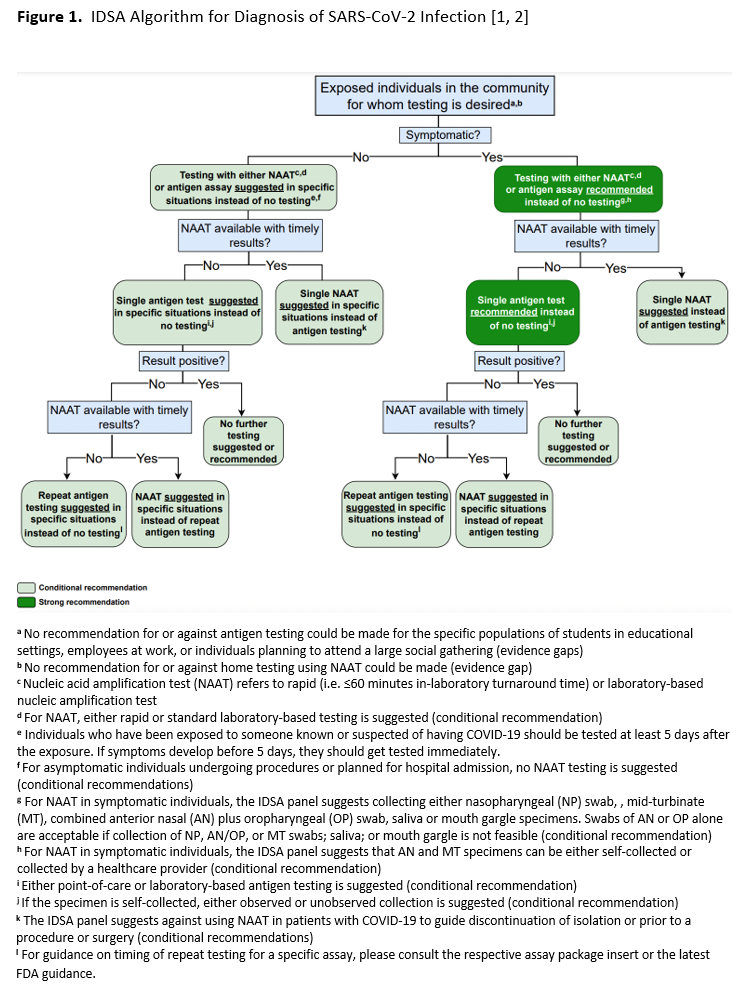
Recommendations
Recommendation 1: The IDSA panel recommends a SARS-CoV-2 NAAT in symptomatic individuals suspected of having COVID-19 (strong recommendation, moderate certainty evidence).
- Remarks:
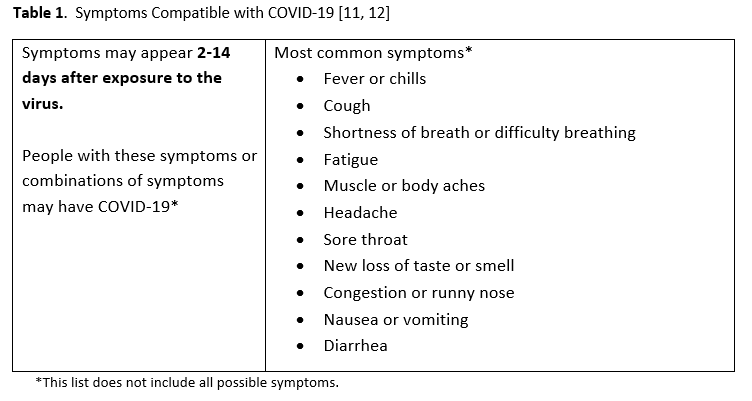
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19 (Table 1).
- Testing is indicated since clinical assessment alone is not accurate in predicting a COVID-19 diagnosis. A positive test result may inform decisions about therapy, isolation, and potentially contact tracing.
- There were limited data available regarding the analytical performance of SARS-CoV-2 NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, in children, or in patients infected with SARS-CoV-2 variants (e.g., Omicron).
Recommendation 2: For symptomatic individuals suspected of having COVID-19, the IDSA panel suggests collecting and testing swab specimens from either the nasopharynx, anterior nares, oropharynx, or mid-turbinate (MT) regions; saliva, or mouth gargle (conditional recommendation, low certainty evidence).
- Remarks:
- Compared to NP swabs, AN or OP swabs yield more false-negative results than combined AN/OP swabs, MT swabs, saliva, or mouth gargle (Table 2). Swabs of AN or OP alone are acceptable if collection of NP, AN/OP, or MT swabs; saliva; or mouth gargle is not feasible.
- Sample collection methods are not standardized (e.g., drool or spit with/without cough were all reported as saliva).
- The patient’s ability to follow instructions and cooperate with requirements of specimen collection (e.g., spit into a container, nothing by mouth for some time before saliva collection) should be considered.
- FDA approval of individual NAATs specifically indicates collection and specimen type(s). Failure to adhere to label requirements, unless otherwise approved through a lab developed test (LDT) validation or authorized by the FDA through a subsequent EUA for different collection or specimen type, can lead to inaccurate results and reimbursement denials.
Recommendation 3: The IDSA panel suggests that for symptomatic individuals suspected of having COVID-19, anterior nasal (AN) and MT swab specimens may be collected for SARS-CoV-2 RNA testing by either patients or healthcare providers (conditional recommendation, moderate certainty evidence).
- Remarks:
- An important limitation of the data available to inform this recommendation is that the type of specimen differed by comparison group. That is, while self-collected samples were always AN and MT specimens, healthcare provider-collected samples were always NP specimens. This might explain the increased sensitivity of healthcare provider collected specimens.
Recommendation 4: The IDSA panel suggests using either rapid or standard laboratory-based NAATs in symptomatic individuals suspected of having COVID-19 (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensuring high-quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60 minutes or even longer. For this guideline, rapid testing was defined as a turnaround time of 60 minutes or less. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid test methodologies include rapid reverse transcription polymerase chain reaction (RT-PCR) and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
- This recommendation applies only to tests evaluated in the included studies. One test, Abbott IDNow, was included in most of the studies evaluated in this recommendation and may have skewed results towards lower sensitivity. Variability of test performance with different specimen-types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with contemporary SARS-CoV-2 variants.
Recommendation 5: The IDSA panel suggests performing a single NAAT and not repeating testing routinely in symptomatic or asymptomatic individuals suspected of having COVID-19 whose initial NAAT result is negative (conditional recommendation, very low certainty of evidence).
- Remarks:
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19 (Table 1).
- While the yield of repeat testing is low and therefore repeat testing is not suggested routinely, there may be situations where repeat testing might be considered. An example of such a situation is the development of new or worsening symptoms in the absence of alternative explanations. Also, timing of symptom onset might drive a need for repeat testing. A poorly collected specimen could yield a falsely negative result and might be another reason supporting repeat testing.
- If performed, repeat testing should generally occur 24-48 hours after initial testing and once the initial NAAT result has returned as negative. Another specimen type, preferably a lower respiratory tract specimen if the patient has signs/symptoms of lower respiratory tract infection, should be considered for repeat testing.
Recommendation 6: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests SARS-CoV-2 RNA testing in asymptomatic individuals who are either known or suspected to have been exposed to COVID-19 (conditional recommendation, moderate certainty evidence).
- Remarks:
- The panel recognizes the lack of evidence supporting therapy for asymptomatic persons and the absence of treatment approved through EUA for asymptomatic COVID-19 but acknowledges that individual clinical scenarios may lead clinicians toward testing and consideration of treatment. Individuals who have clinical or epidemiologic reasons that might make testing desirable (e.g., high-risk individuals, such as those who have pulmonary conditions or are immunocompromised or those in close contact with immunocompromised individuals) may be considered for testing. Testing should be done at least 5 days after the exposure. If symptoms develop before 5 days, the exposed individual should get tested immediately. Knowledge that an individual is infected with SARS-CoV-2 can be helpful to inform appropriate isolation [3]. The decision to test asymptomatic persons should depend on the availability of testing resources. Known exposures are defined herein as close contact for at least 15 minutes over a 24-hour period with someone who has laboratory-confirmed COVID-19. Suspected exposures might be defined as working or residing in a congregate setting (e.g., long-term care or correctional facility, cruise ship, factory) experiencing a COVID-19 outbreak. The risk of contracting SARS-CoV-2 may vary under different exposure conditions, e.g., length of time exposed, indoor versus outdoor setting, whether masks were worn routinely. Household contacts may be especially high-risk. This recommendation assumes the exposed individual was not wearing appropriate PPE.
Recommendation 7: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests using either rapid or standard laboratory-based NAATs in asymptomatic individuals with known exposure to SARS-CoV-2 infection (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensure quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60 minutes or even longer. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid test methodologies include rapid RT-PCR and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
- This recommendation applies only to tests evaluated in the included studies. One test, Abbott IDNow, was included in most of the studies evaluated in this recommendation and may have skewed results towards lower sensitivity. Variability of test performance with different specimen types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with different SARS-CoV-2 variants.
Recommendation 8: The IDSA panel suggests against routine SARS-CoV-2 NAAT in asymptomatic individuals without a known exposure to COVID-19 who are being hospitalized (conditional recommendation, very low certainty evidence).
- Remarks:
- Important considerations for this recommendation are that the IDSA panel was unable to identify studies published during the period of literature review that showed reduced SARS-CoV-2 transmission to healthcare providers or to other patients resulting from prehospitalization testing. The evidence was indirect and assessed only diagnostic test accuracy in studies of symptomatic patients alone or together with asymptomatic patients. The burden of testing all patients planned to be admitted was considered, in the face of limited evidence. Finally, there are other effective infection prevention interventions, including use of PPE and vaccination that should be considered.
- The panel acknowledges that there could be a benefit of pre-admission NAAT in some situations, such as admission to a multibed room; to a unit with a congregate treatment area, such as a behavioral health unit; or to a positive pressure room or unit.
- This recommendation addresses only acute care hospital settings.
Recommendation 9: The IDSA panel suggests against routine SARS-CoV-2 NAAT of asymptomatic individuals without a known exposure to COVID-19 who are undergoing a medical or surgical procedure (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on non-standardized instrument signal values, such as cycle threshold (Ct) values.
- Decisions on the timing of a procedure in a patient with prior SARS CoV-2 infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age), procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
- Given limited evidence for poor outcomes in asymptomatic persons who undergo major surgery soon after testing positive for SARS-CoV-2 infection, testing may be considered during periods of high community transmission.
- Testing may also be considered before solid organ transplantation, hematopoietic stem cell transplantation or CAR-T cell therapy.
- This recommendation applies to settings where protective measures, such as PPE, are available and are used with adherence. Other factors to consider include the vaccination status of healthcare providers and patients, and whether patients will be roomed with other patients before or after the procedure. This recommendation is based on general exposure in the community as compared to a specific known exposure.
Recommendation 10: The IDSA panel suggests against routinely repeating NAAT before medical or surgical procedures in patients with a recent history of COVID-19 (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
- Decisions on the timing of a procedure in a patient with prior SARS CoV-2 infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age) procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
Recommendation 11: The IDSA panel suggests against routinely repeating NAAT in patients with COVID-19 to guide release from isolation (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens for prolonged periods without evidence of infectious virus has been widely reported. Predicating release from isolation on a negative SARS-CoV-2 NAAT may unnecessarily extend the duration of isolation.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
Recommendation 12: The IDSA panel suggests neither for nor against home testing for SARS-CoV-2 (evidence gap).
Background
In late December 2019, an outbreak of pneumonia cases of unclear etiology was reported in Wuhan City, Hubei Province, China [4]. Unbiased next generation sequencing (NGS) using lower respiratory tract (LRT) specimens collected from affected patients subsequently identified a novel coronavirus as the cause of illness now known as Coronavirus Disease 2019 (COVID-19). The entire viral genome was shared online within days and phylogenetic analyses established close relationship to human severe acute respiratory syndrome coronavirus (SARS-CoV) as well as several other SARS-like bat coronaviruses [4, 5]. Based on genetic similarities, the novel coronavirus was officially named SARS-CoV-2 [6]. By March 11th, 2020, the virus had spread to at least 114 countries and killed more than 4,000 people, prompting the World Health Organization (WHO) to officially declare a global pandemic [7].
Public availability of the SARS-CoV-2 genome was an essential first step enabling development of accurate molecular diagnostic assays. Nucleic acid amplification tests designed to detect one or more gene sequences specific to SARS-CoV-2 are essential for confirming COVID-19 diagnoses. On February 4, 2020, the United States (U.S.) Secretary of Health and Human Services announced that circumstances existed justifying authorization of the emergency use of SARS-CoV-2 molecular tests [8]. This declaration meant that commercial manufacturers and clinical laboratories were required to submit details about their SARS-CoV-2 assays to the U.S. FDA for review and Emergency Use Authorization (EUA).
To date, multiple commercial test manufacturers and clinical laboratories, including academic medical centers, have received EUA for a SARS-CoV-2-specific molecular diagnostic tests, including direct to consumer and over the counter tests manufactured for home use [9]. One multiplex NAAT has received full FDA approval under the traditional premarket review process [10]. It is important to recognize that EUA guidance differs from the standard FDA approval process. In the setting of a public health emergency, the FDA only requires test developers to establish acceptable analytical accuracy. Clinical test performance (i.e., clinical sensitivity and specificity) has yet to be determined or comprehensively compared across EUA platforms.
Given increasing test availability combined with a rapidly growing number of NAAT-focused studies published in academic journals, IDSA formed a multidisciplinary panel to critically appraise the existing literature and develop evidence-based diagnostic test recommendations. The panel identified and prioritized practical diagnostic questions pertaining to symptomatic patients and asymptomatic individuals to drive the literature review. The symptoms considered compatible with COVID-19 are listed in Table 1.
It is anticipated that these guidelines will continue to be updated as substantive new information becomes available.
Methods and Search Results
The guideline was developed using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach for evidence assessment. In addition, given the need for rapid response to an urgent public health crisis, the methodological approach was modified according to the Guidelines International Network/McMaster checklist for development of rapid recommendations [13]. This guideline serves as the second update to the original IDSA Guidelines on the Diagnosis of COVID-19 [1]. This update addresses 8 questions that were addressed previously and 4 new questions that were considered clinically important. The list of questions can be found under Table s1.
Panel Composition
The panel was composed of eight members including frontline clinicians, infectious diseases specialists, healthcare epidemiologists and clinical microbiologists who were members of IDSA, American Society for Microbiology (ASM), Society for Healthcare Epidemiology of America (SHEA), and the Pediatric Infectious Diseases Society (PIDS). They represented the disciplines of adult and pediatric infectious diseases, medical microbiology, as well as nephrology and gastroenterology. The Evidence Foundation provided technical support and guideline methodologists for the development of this guideline.
Disclosure and Management of Potential Conflict of Interest
The conflict of interest (COI) review group included two representatives from IDSA who were responsible for reviewing, evaluating, and approving all disclosures. All members of the expert panel complied with the COI process for reviewing and managing conflicts of interest, which required disclosure of any financial, intellectual, or other interest that might be construed as constituting an actual, potential, or apparent conflict, regardless of relevancy to the guideline topic. The assessment of disclosed relationships for possible COI was based on the relative weight of the financial relationship (i.e., monetary amount) and the relevance of the relationship (i.e., the degree to which an association might reasonably be interpreted by an independent observer as related to the topic or recommendation of consideration). The COI review group ensured that the majority of the panel and chair was without potential relevant (related to the topic) conflicts. The chair and all members of the technical team were determined to be unconflicted.
Question Generation
For the original guideline, clinical questions were developed into a Population, Intervention, Comparison, Outcomes (PICO) format [14] prior to the first panel meeting (Table s1). IDSA panel members prioritized questions with available evidence that met the minimum acceptable criteria (i.e., the body of evidence reported on at least test accuracy results can be applied to the population of interest). Panel members prioritized patient-oriented outcomes related to SARS-CoV-2 testing, such as requirement for self-quarantine, eligibility for COVID-19 treatment, and timing of elective surgery or procedures. We also considered the impact of SARS-CoV-2 results on infection prevention and public health practices, including the use of PPE and contact tracing. In this update, the panel retired the question related to testing specimens from the upper versus lower respiratory tract in patients with lower respiratory tract disease, and the question related to testing of immunocompromised individuals, due to absence of relevant data. The third version of the guideline attempted to focus on clinically relevant questions for which new data might be available to inform a new recommendation or to change the direction or strength of an earlier recommendation. The panel also addressed questions within its scope of expertise (e.g. avoided questions that required expertise in immunology). (See Table s1)
Search Strategy
The National Institute of Health and Care Excellence (NICE) and the Center for Disease Control and Prevention (CDC) highly sensitive search was reviewed by the methodologist in consultation with the technical team information specialist and was determined to have high sensitivity. An additional term, COVID, was added to the search strategy used in addition to the terms identified in the PICO questions (Table s2). Ovid Medline, Embase, and Cochrane databases were searched for studies from 2019 through July 1, 2022. We also performed horizon scans and consulted with field experts during the evidence assessment and recommendation process to locate additional literature. This was especially relevant to answer PICO questions for which test accuracy results were not available. Preprints were followed for final publication but preprints were not included in the literature review unless published, since the reviewer team identified a sufficient number of published studies. Reference lists and literature suggested by panelists were reviewed for inclusion.
Screening and Study Selection
A member of the reviewer team screened titles and abstracts, as well as eligible full-text studies. We included studies reporting data on diagnostic test accuracy (cohort studies, cross sectional studies and case-control studies). When questions compared the performance of different tests (e.g., different testing or sampling methods) or testing strategies, we included studies that provided direct test accuracy data about all tests in the same population, referred to as direct comparative test accuracy studies. For this analysis, studies were excluded if all patients did not receive all tests. When these direct studies where lacking, we included studies that assessed a single test and compared its results to a reference standard.
We excluded studies of index tests that did not have EUA or CE status, and tests for which EUA/CE status could not be confirmed due to a lack of reporting of test name, studies with fewer than 30 patients/samples, machine learning studies, protocols, studies with incomplete test accuracy information (i.e., reported sensitivity without specificity), studies reporting atypical sample site collection (e.g., wastewater, conjunctival swab, fecal/ anal swab, seminal fluid, peritoneal swab, environmental surfaces, air samples, breath condensate), studies that used uncommonly available assays (i.e., CRISPR-Cas9 genome editing, bioelectric Recognition Assay (BERA), attenuated total reflection-Fourier-transform infrared (ATR-FTIR) spectroscopy), and studies that that compared two RT-PCR component kits (probes, primers, enzymes, swab type, master mix). We presumed patients to be truly positive for SARS-CoV-2 infection if an assay provided a positive result for at least one target gene.
For the direct comparative test accuracy studies (including rapid versus standard tests), data were abstracted with each test as the index test and the combination of tests as a reference standard. The panel determined the combination of tests reference standard would be a minimum of at least positive tests. For example, if one of four tests were positive, this patient would be considered negative. If two out four tests were positive, this patient would be considered positive. For some of comparisons, we established a different reference standard to reduce potential bias. For example, with the comparison of self and healthcare provider sample collection, any single positive result was considered the reference standard.
That is, if the sample was positive by self-collection only, it was considered positive and became part of the total positive samples for sensitivity calculation.
Data Collection and Analysis
A member of the review team completed data extraction using a standardized data extraction form. The IDSA panel members served as a second reviewer for results relating to sample site collection, with no major discrepancies found. Data extracted included general study characteristics (authors, publication year, country, study design), diagnostic index test and reference standard, prevalence of SARS-CoV-2 infection, and parameters to determine test accuracy (i.e., sensitivity and specificity of the index test).
For each study, we calculated the sensitivity and specificity of the diagnostic index test and used the Clopper–Pearson method to estimate 95% confidence intervals. We then fit the random-effects bivariate binomial model of Chu and Cole (1) to pool accuracy estimates using the glmer function of the lme4 package in R (version 4.1.2). To pool accuracy estimates for analyses including fewer than5 studies, we fit a fixed effects model as implemented in the meta package in R (version 4.1.2). We used forest plots to plot individual and summary estimates and conducted subgroup analyses to explore heterogeneity.
To calculate the absolute differences in effects for different testing or sampling strategies, we applied the results of the sensitivity and specificity to a range of plausible prevalence in the population. We then calculated true positives, true negatives, false positives, and false negatives. To determine the prevalence for each question, we considered the published literature in consultation with the clinical experts. Prevalence, defined by the results of surveillance testing in a community, has been shown to change over time. For the purposes of the guideline, we used 1%, 5%, and 10% pre-test probability to mirror a range of community prevalence in asymptomatic persons.
Risk of Bias and Certainty of Evidence
We conducted the risk of bias assessment for diagnostic test accuracy studies using the Quality Assessment of Diagnostic Accuracy Studies (QUADAS)-2 revised tool (Table s3) [15]. GRADE framework was used to assess overall certainty by evaluating the evidence for each outcome on the following domains: risk of bias, imprecision, inconsistency, indirectness, and publication bias [16, 17]. GRADE summary of findings tables were developed in GRADEpro Guideline Development Tool [18].
Evidence to Recommendations
The panel considered core elements of the GRADE evidence in the decision process, including certainty of evidence and balance between desirable and undesirable effects. Additional domains were acknowledged where applicable (e.g., feasibility, resource use, acceptability). For all recommendations, the expert panelists reached consensus. Voting rules were agreed on prior to the panel meetings for situations when consensus could not be reached.
As per GRADE methodology, recommendations are labeled as “strong” or “conditional”. The words “we recommend” indicate strong recommendations and “we suggest” indicate conditional recommendations. Figure 2 provides the suggested interpretation of strong and weak recommendations for patients, clinicians, and healthcare policymakers. Rarely, low certainty evidence may lead to strong recommendations. In those instances, we followed generally recommended approaches by the GRADE working group, which are outlined in five paradigmatic situations (e.g., avoiding a catastrophic harm) [19]. For recommendations pertaining to good practice statements, appropriate identification and wording choices were followed according to the GRADE working group [20]. A “good practice statement” represents a message perceived by the guideline panel as necessary to health care practice, that is supported by a large body of indirect evidence difficult to summarize and indicates that implementing this recommendation would clearly result in large net positive consequences. For recommendations where the comparators are not formally stated, the comparison of interest was implicitly referred to as “not using the test”. Some recommendations acknowledge the current “knowledge gap” and aim at avoiding premature favorable recommendations for test use and to avoid encouraging the rapid diffusion of potentially inaccurate tests.
Revision Process
The draft guideline underwent rapid review for approval by IDSA Board of Directors Executive Committee external to the guideline development panel. The guideline was reviewed by ASM, SHEA and PIDS, and endorsed by ASM and PIDS. The IDSA Board of Directors Executive Committee reviewed and approved the guideline prior to dissemination.
Revision Process
The draft guideline underwent rapid review for approval by IDSA Board of Directors Executive Committee external to the guideline development panel. The guideline was reviewed by ASM, SHEA and PIDS, and endorsed by all three societies. The IDSA Board of Directors Executive Committee reviewed and approved the guideline prior to dissemination.
Updating Process
Regular, frequent screening of the literature will take place to determine the need for revisions based on the likelihood that new data will have an impact on the recommendations. If necessary, the expert panel will be reconvened to discuss potential changes.
Search Results
The systematic review search across the three databases identified 22,586 references. After screening titles and abstracts, 1,275 references were marked to answer the molecular PICO questions. Of those, 88 studies answered at least one of the 12 PICO questions. 15 additional studies were incorporated from targeted searches for PICO questions that lacked test accuracy studies, bringing the total to 103 studies informing this guideline update (Figure s1). Characteristics of the included studies can be found in Table s4. Narrative summaries of the additional studies can be found in Table s5.
Recommendation 1: NAAT in Symptomatic Individuals
Recommendation 1: The IDSA panel recommends a SARS-CoV-2 NAAT in symptomatic individuals suspected of having COVID-19 (strong recommendation, moderate certainty evidence).
- Remarks:
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19 (Table 1).
- A positive test result may inform decisions about therapy, isolation, and potentially contact tracing.
- There were limited data available regarding the analytical performance of SARS-CoV-2 NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, in children, or in patients infected with recent SARS-CoV-2 variants (e.g., Omicron).
Summary of the evidence
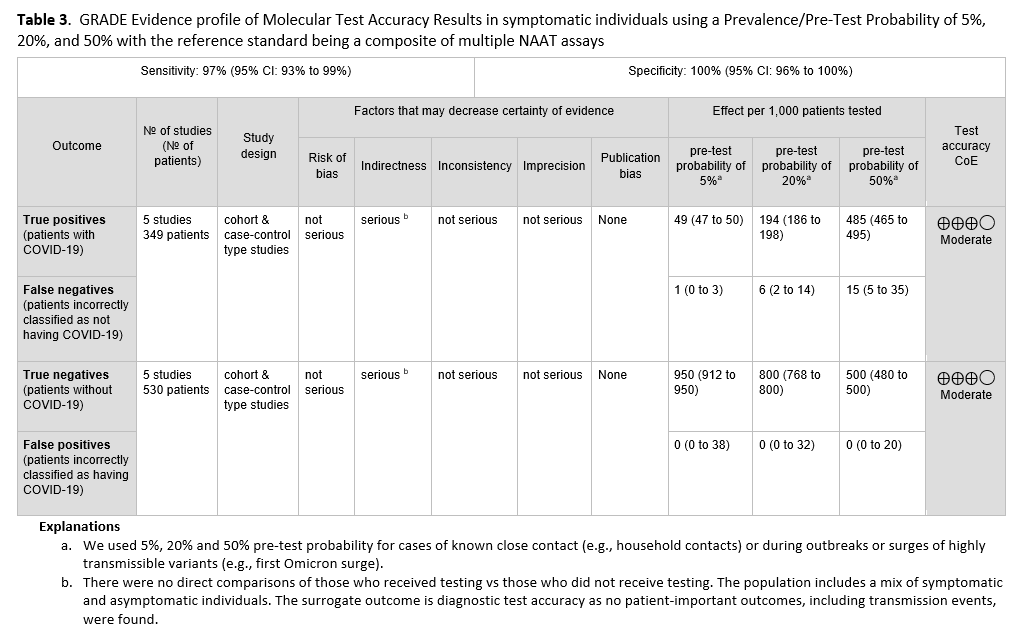
No direct evidence that assessed patient- or population-centered outcomes of testing versus no testing in symptomatic patients was found. Therefore, the panel relied on diagnostic test accuracy data to inform this recommendation. Five studies [21-25] with 349 positive and 530 negative patients, based on standard NAAT, were used to inform this recommendation (See Figures s2a-s2b). The reference standard was a composite of more than two standard NAATs (i.e., rapid RT-PCR and laboratory-based NAAT) using NP or nasal swabs. The pooled sensitivity was 97% (95% CI: 93 to 99) and the pooled specificity was 100% (95% CI: 96 to 100). The IDSA panel considered prevalences of 5%, 20% and 50% in symptomatic patients (i.e., those with at least one of the common symptoms of COVID-19) (See Table 2). Across this range of prevalences, there were 1-15 predicted false-negative results and no predicted false-positive results per 1,000 individuals (See Table 3). The certainty of the evidence is moderate due to rating downward for indirectness. The indirectness was due to lack of direct comparisons of those who received testing versus those who did not receive testing. Also, most studies reported results of testing a mixed population of symptomatic and asymptomatic individuals or did not specify the symptom status of the population tested. This added to the indirectness, as the question addresses symptomatic individuals.
Benefits and harms
Diagnostic testing for symptomatic SARS-CoV-2 infection is needed because clinical assessment alone is not accurate in predicting a COVID-19 diagnosis. Some infected individuals may incorrectly believe that since they have already been infected with SARS-CoV-2 and/or been vaccinated, they do not need testing because they are not at risk for SARS-CoV-2 infection; such individuals may not take appropriate infection prevention precautions, which could lead to spread of SARS-CoV-2 or they may not obtain medical follow-up or receive needed treatment unless tested. Similarly, false-negative results are a potential harm of testing; false-negative test results could cause symptomatic individuals to spread SARS-CoV-2 and deny such patients appropriate therapy. The potential harm of false-positive results includes isolating individuals who do not have COVID-19, causing unnecessary anxiety, delaying additional evaluation to look for the cause of symptoms, administering unnecessary therapeutics for COVID-19, increasing days away from work or school, and unnecessary contact tracing. The very high specificity of standard NAAT (i.e., no predicted false positives at prevalences ranging from 5% to 50%) minimizes these potential harms.
Additional considerations
SARS-CoV-2 testing is generally acceptable to patients and providers. In the U.S., individuals with health insurance should not pay out-of-pocket costs for COVID-19 testing. NAAT can also be accessed through programs in some communities at no-cost. This may change in the future and may impact the likelihood of patients seeking testing.
Conclusions and research needs for this recommendation
SARS-CoV-2 NAAT testing is recommended for symptomatic individuals in the community with a compatible clinical syndrome (Table 1).
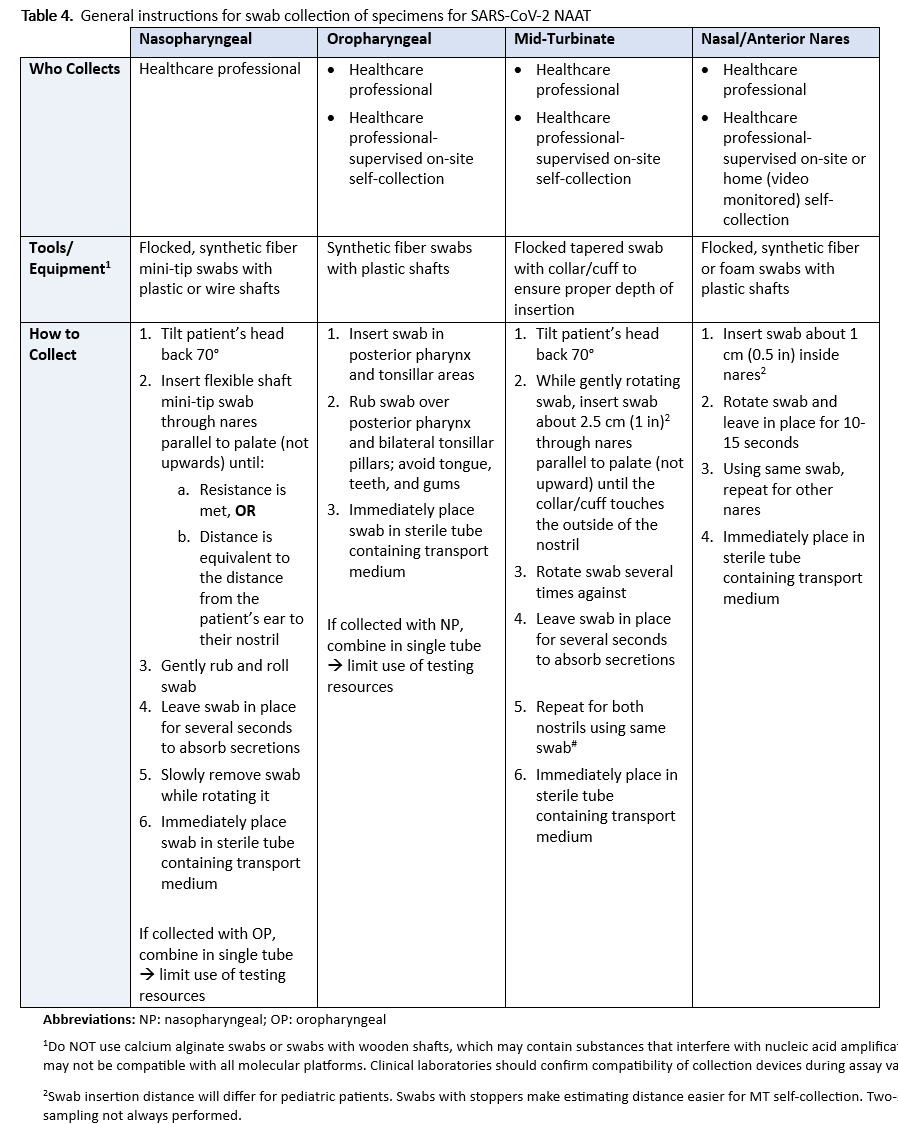
Recommendation 2: Anatomic Site of Specimen Collection
Recommendation 2: For symptomatic individuals suspected of having COVID-19, the IDSA panel suggests collecting and testing swab specimens from either the nasopharynx (NP), anterior nares (AN), oropharynx (OP), or midturbinate regions (MT); saliva, or mouth gargle (conditional recommendation, low certainty evidence).
- Remarks
- Compared to NP swabs, AN or OP swabs alone yield more false-negative results than combined AN/OP swabs, MT swabs, saliva, or mouth gargle (Table 2). Swabs of AN or OP alone are acceptable if collection of NP, AN/OP, or MT swabs, saliva, or mouth gargle is not feasible.
- Sample collection methods are not standardized (e.g., drool or spit with/without cough were all reported as saliva)
- The patient’s ability to follow instructions and cooperate with requirements of specimen collection (e.g., spit into a container, nothing by mouth for some time before saliva collection) should be considered.
- FDA approval of individual NAATs specifically indicates collection and specimen type(s). Failure to adhere to label requirements, unless otherwise approved through a lab developed test (LDT) validation or authorized by the FDA through a subsequent EUA for different collection or specimen type, can lead to inaccurate results and reimbursement denials.
Summary of the evidence
We reviewed the published literature to identify studies assessing the performance of different specimen types relative to NP swabs for detection of SARS-CoV-2 RNA. Specimen types were grouped into NP swabs, MT swabs, AN swabs, or oropharyngeal (OP) swabs (also referred to as “throat” swabs), saliva, mouth gargle (also referred to as oral rinse, mouth lavage, mouthwash, saline gargle) or a combined swab sampling of AN and OP. A swab insertion cutoff of 0.5 inch was used to define an AN specimen and to differentiate this specimen type from MT. Due to variability in collection methods, saliva specimens were further subdivided into saliva with coughing and saliva without coughing, depending upon whether the study methodology included asking individuals to cough prior to saliva collection.
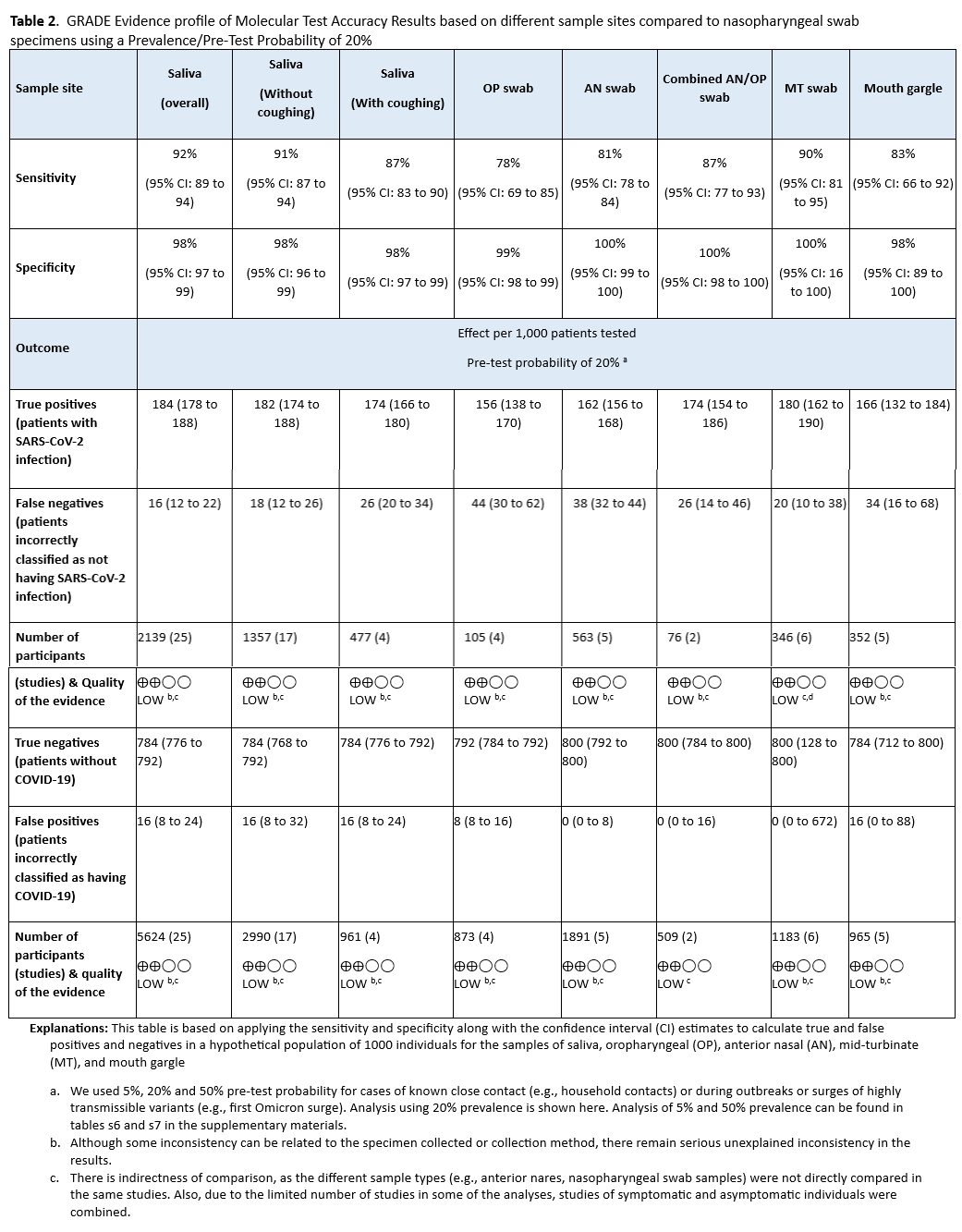
Forty-four studies [28-70] reported the test accuracy of different specimen types using a NP swab as the reference, with the sensitivity ranging between 78% (95% CI: 69% - 85%) for OP to 92% (95% CI: 89% - 94%) for saliva overall. (See Figures s3a till s11b). Sensitivity for OP and AN (81%, 95% CI: 78-84%) swabs alone were lower than for the other specimen types, with a corresponding increase in false-negative results (Table 2, Tables s6-s7). The specificity for all specimen types ranged from 98% to 100%. Saliva was the most frequently assessed index specimen type. (See Table 2, Tables s6-s7).
The quality of the evidence varied for different specimen sites and was lowered for indirectness, inconsistency, and imprecision. Indirectness was related to comparison (each sample site was from a different population) and to population (several studies included a mix of symptomatic and asymptomatic individuals). Inconsistent results for testing of specimens from some of the same sample sites remained unexplained even after critical review of outlier studies. The low number of patients included in studies resulted in lowering for imprecision in some of the analyses.
Benefits and harms
The panel recognizes that analytical sensitivity may be lower with some alternative specimen types such as AN, OP compared to NP swab (See Table 2), although reports are mixed. For example, a study in a pediatric population that was published after the completion of the literature search for this guideline found similar SARS-CoV-2 Ct value distributions for AN and NP swab samples from symptomatic children during the Delta and initial Omicron surges, suggesting that sensitivity of these two sample types may be comparable for more recent virus variants [71]. The NP swab collection is more invasive and uncomfortable compared to the other studied specimen collection methods and requires a trained healthcare provider to collect. Simple, non-invasive methods of specimen collection that require minimal healthcare provider training and time and that include the possibility of self-collection may result in more testing being done. In developing this recommendation, the panel concluded that the benefit of increased testing outweighed the potential harm of a higher number of false-negative results.
Additional considerations
There are inherent challenges in collecting upper respiratory specimens, including anatomic sites of shedding, consistent swabbing techniques, and in cases of saliva collection, time from eating/drinking, use of coughing and other variables such as mouthwash. While NP swab collection has become the preferred reference technique, there is no true gold standard to compare NP against alternative specimens like AN, MP, OP, saliva, or mouth gargle. Additionally, the actual collection varies considerably; see Table 4 for examples of specimen collection instructions Unlike a blood draw or urine collection, swab collection of the upper respiratory tract is not uniform. Therefore, there is uncertainty about whether the specimen is optimal even with NP collections. Lastly, FDA approval of individual NAATs, including multiplex assays that also test for other respiratory pathogens, specifically indicates collection and specimen type(s). Failure to adhere to label requirements, unless otherwise approved through a lab developed test (LDT) validation or authorized by the FDA through a subsequent EUA for different collection or specimen type, can lead to inaccurate results and reimbursement denials.
Conclusions and research needs for this recommendation
For community and outpatient testing, the benefits support the use of alternative collection methods and specimen types. Studies comparing NP against alternative upper respiratory collection methods using a more comprehensive reference method are needed.
Recommendation 3: Self-Collection of Swab Specimens
Recommendation 3: The IDSA panel suggests that for symptomatic individuals suspected of having COVID-19, AN and MT swab specimens may be collected for SARS-CoV-2 RNA testing by either patients or healthcare providers (conditional recommendation, moderate certainty evidence).
- Remarks:
- An important limitation of the data available to inform this recommendation is that the type of specimen differed by comparison group. That is, while self-collected samples were always AN and MT specimens, healthcare provider-collected samples were always NP specimens. This might explain the increased sensitivity of healthcare provider collected specimens.
Summary of the evidence
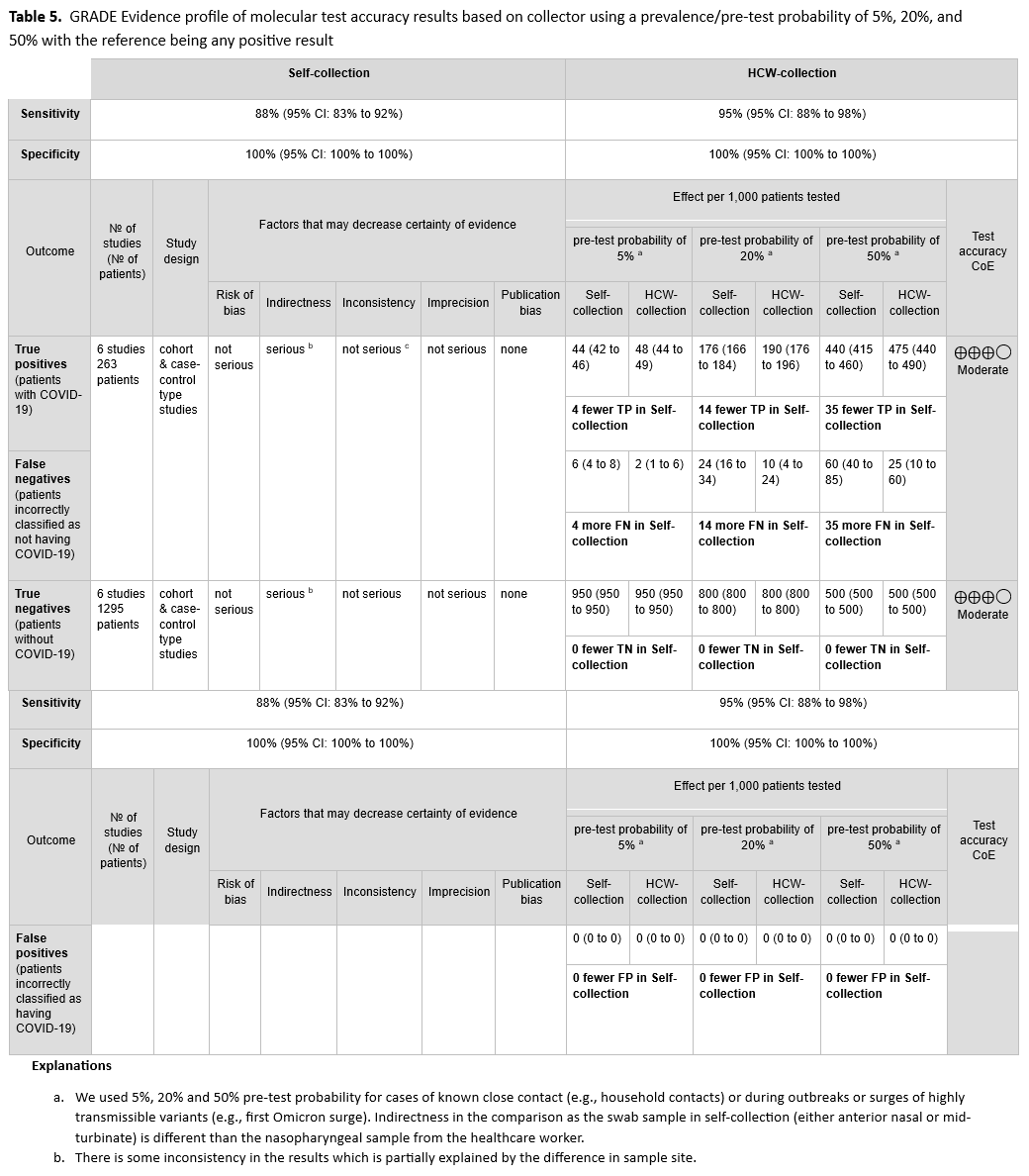
We identified six cohort studies with 263 positive and 1295 negative patients that reported results of self-collected samples compared to healthcare provider collected samples. [38, 59, 64, 65, 72, 73]. (See Figures s12a till s13b). The self-collected sample sites were AN and MT. We excluded studies in which sample collection was not done with a swab (i.e., saliva, mouth gargle, sputum). In this analysis, we did not consider the healthcare provider collection as the reference standard, instead, any positive test whether self-collected or healthcare provider collected was considered to be a true positive. The pooled sensitivity of self-collected AN or MT specimens was 88% (95% CI: 83% to 92%), and the pooled sensitivity of healthcare provider collected NP specimens was 95% (95% CI: 88% to 98%). (See Table 5)
The certainty of the evidence was rated low due to indirectness of comparisons and inconsistency. In all six studies, the healthcare provider collected a NP specimen, whereas all self-collections were either AN or MT specimens. The unexplained inconsistency of results of testing the same specimen type also further lowered the certainty of the evidence to low.
Benefits and harms
Benefits to self-collected specimens include convenience for patients, including the possibility of home collection, and a reduced burden on healthcare providers and other healthcare resources. The panel assumes the importance of individuals who self-collect specimens to be well-trained to follow step-by-step protocols, although the impact of training on quality of specimen collection was not assessed. Educational materials and easy to follow instructions, including in multiple languages, are assumed to be useful, although again, this was not assessed specifically when developing this recommendation.
Additional considerations
In a study published after this guideline’s evidence review cutoff date, children as young as kindergarten age were able to successfully self-collect AN specimens for SARS-CoV-2 NAAT; the positive and negative percent agreement compared with health care provider-collected anterior nares specimens were 97.8% (95% CI, 94.7%-100.0%) and 98.1% (95% CI, 95.6%-100.0%), respectively. This provides support for the hypothesis that the greater sensitivity of healthcare provider collected specimens compared to self-collected specimens in the studies included in the literature search for this recommendation may have been due in large part to specimen source (i.e., NP swab collected by healthcare provider versus AN swab collected by patient). It should be noted as well that most self-collection studies were performed in the presence of a healthcare provider. The studies were based on symptomatic patients so data on self-collection in asymptomatic individuals are lacking. Additionally, not all assays have FDA indications for self-collection.
Conclusions and research needs for this recommendation
Self-collection of swab specimens from symptomatic individuals in the community who are suspected of having COVID-19 has similar diagnostic accuracy to healthcare provider-collected specimens. Studies in which self-collection is done without the supervision of healthcare providers and in asymptomatic individuals are needed.
Recommendation 4: Rapid versus Standard Laboratory-based NAATs in Symptomatic Individuals
Recommendation 4: The IDSA panel suggests using either rapid or standard laboratory-based NAATs in symptomatic individuals suspected of having COVID-19 (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensuring high-quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60-minutes or even longer. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid molecular test methodologies include rapid reverse transcription polymerase chain reaction (RT-PCR) and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
- This recommendation applies only to tests evaluated in the included studies. One test, Abbott IDNow, was included in most of the studies evaluated in this recommendation and may have skewed results towards lower sensitivity. Variability of test performance with different specimen-types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with contemporary SARS-CoV-2 variants.
Summary of the evidence
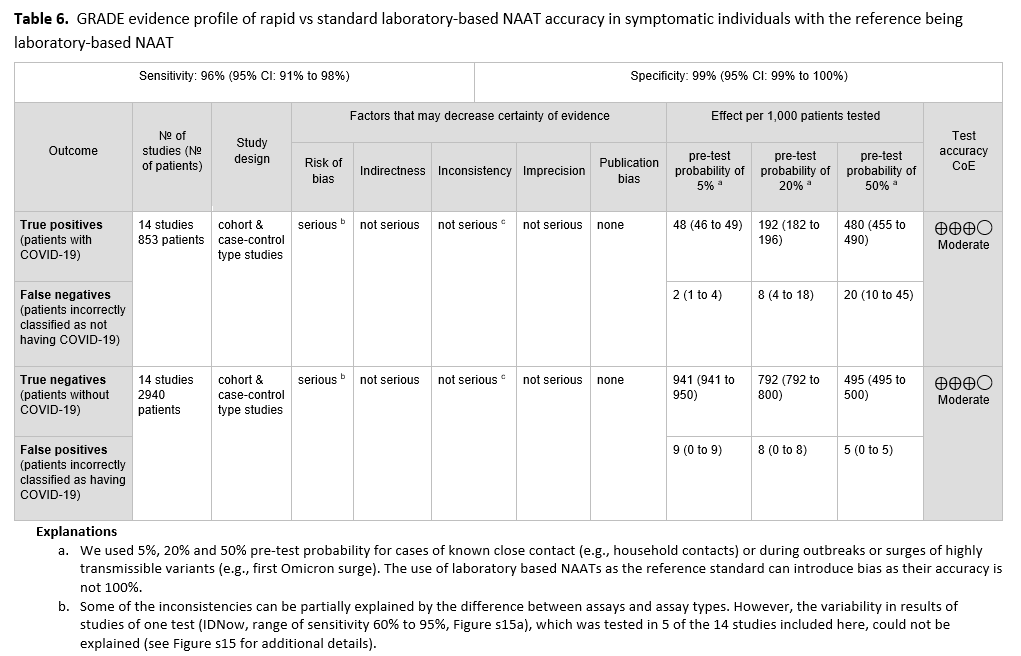
Fourteen studies [74-87] of rapid and laboratory based molecular diagnostic tests that included 853 patients with a positive result and 2,940 patients with a negative result informed this recommendation. The reference test was a laboratory based NAAT. The pooled sensitivity of the rapid NAATs evaluated was 96% (95% CI: 91% to 98%) and the pooled specificity was 100% (95% CI: 98% to 100%). (See Figures s14a till s15b). We considered 5%, 20% and 50% as prevalences (pre-test probabilities) of COVID-19 in symptomatic patients (i.e., those with at least one common symptom of COVID-19). Across the prevalences considered, false negative results were predicted to range from 2 to 20 and false positive results to range from 5 to 9 per 1000 patients tested. The certainty of the evidence was moderate due to risk of bias with the use of laboratory-based NAAT as the reference standard, and with some inconsistency of sensitivity results for some assays. (See Table 6).
Benefits and harms
The value of rapid molecular diagnostic testing, ideally completed while the patient is physically present, is that it allows management decisions related to treatment and isolation to be made and enacted quickly. Rapid testing may reduce the period of patient anxiety while test results are pending. A rapid result decreases concerns about losing patients to follow-up and generally simplifies follow-up.
An important finding here is that rapid diagnostic tests can yield accurate results. All rapid molecular tests had very high specificities, minimizing false positive results. The potential harms of false positive results include isolating individuals who do not have COVID-19 infection, causing unnecessary anxiety, delaying additional investigation for the true cause of symptoms, administering unnecessary therapy for COVID-19, increasing days away from work or school, and unnecessary contact tracing. Positive results from rapid NAATs do not usually need to be confirmed by standard laboratory-based NAATs. In addition, the pooled sensitivity of the rapid tests was 96% implying that rapid molecular tests, especially RT-PCR tests, perform as well as standard laboratory-based molecular tests. This indicates that negative rapid molecular test results do not need routine confirmation.
Additional considerations
Timing of testing relative to symptom onset may impact sensitivity of rapid and standard laboratory-based NAATs. There were limited data on the performance of rapid NAATs in children. In addition, the specific test, anatomic site, and quality of specimen collection, and use or non-use of viral transport media may affect test performance, regardless of whether it is a rapid or standard NAAT. Lastly, there were no studies directly comparing rapid isothermal NAAT and rapid RT-PCR tests to one another.
Conclusions and research needs for this recommendation
The specificity and sensitivity of rapid and standard laboratory based NAATs appear to be equivalent. Rapid NAATs for SARS-CoV-2 and other microorganisms are an important advance in healthcare and open the door to new ways of healthcare delivery.
Recommendation 5: Single versus Repeat NAAT
Recommendation 5: The IDSA panel suggests performing a single NAAT and not repeating testing routinely in symptomatic or asymptomatic individuals suspected of having COVID-19 whose initial NAAT result is negative (conditional recommendation, very low certainty of evidence).
- Remarks:
- The panel considered symptomatic patients to have at least one of the most common symptoms compatible with COVID-19 (Table 1).
- While repeat testing when the initial test result is negative is not suggested routinely, there may be situations where repeat testing might be considered. An example of such a situation is the development of new or worsening symptoms compatible with COVID-19 in the absence of an alternative explanation. Also, timing of symptom onset might drive a need for repeat testing. A poorly collected specimen could yield a falsely negative result and might be another reason for repeat testing.
- If performed, repeat testing should generally occur 24-48 hours after initial testing and once the initial NAAT result has returned as negative.
Summary of the evidence
These recommendations were based on two studies in which 57 patients tested positive and 2,041 patients tested negative [88, 89]. In these reports, NAAT was performed and repeated using NP swabs collected from symptomatic patients and patients with unreported symptom status. For those undergoing repeat testing, a range of 2 to 3.5% tested positive within one week of an initially negative test. No studies assessing benefits and harms of repeat testing on individual patients or population outcomes were identified. Given the lack of a direct assessment of the implications of single versus repeat testing, the panel assessed the overall certainty of the evidence as being very low.
Benefits and harms
Patients with COVID-19 and a false negative NAAT result may not receive beneficial therapy. Symptomatic or asymptomatic individuals inappropriately labeled as not having COVID-19 may pose a risk of transmitting SARS-CoV-2 to uninfected individuals in the community, healthcare facilities, or congregate settings. The potential harms of false-positive results include isolating individuals who do not have COVID-19, causing unnecessary anxiety, delaying additional evaluation to look for the cause of symptoms, administering unnecessary therapeutics for COVID-19, increasing days away from work or school, and unnecessary contact tracing. Repeat molecular testing may sometimes be warranted to guide treatment or isolation of individuals when the pre-test likelihood of COVID-19 is intermediate or high, but the initial NAAT is negative.
Additional considerations
Consideration of disease prevalence is important given that the negative predictive value of a diagnostic test increases as disease prevalence decreases. Thus, a single negative COVID-19 test result in areas of low disease prevalence is more predictive than in areas of high disease prevalence. Various assays were used in the reported studies and are in use in practice; it was assumed that the performance of all assays was comparable. Only NP swabs were studied and the generalization of these findings to other specimen-types was not directly assessed. The diagnostic yield of a second test may be affected by the duration of symptoms and the anatomic site sampled, including lower respiratory tract specimens.
Conclusions and research needs for this recommendation
Repeat SARS-CoV-2 RNA testing within 7 days of a negative test result rarely yields a positive result, but the evidence supporting this conclusion is of very low quality. When repeat testing is considered, the site and quality of specimen collection should be carefully considered. Further studies evaluating the potential benefit and timing of repeat testing relative to symptom onset in both inpatient and outpatient settings are warranted, as are studies to determine the value of repeating testing on specimen types other than NP swabs.
Recommendation 6: NAAT in Asymptomatic Individuals who are Exposed to SARS-CoV-2
Recommendation 6: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests SARS-CoV-2 RNA testing in asymptomatic individuals who are either known or suspected to have been exposed to COVID-19 (conditional recommendation, moderate certainty evidence).
- Remarks:
- The panel recognizes the lack of evidence supporting therapy for asymptomatic persons and the absence of treatment approved through EUA for asymptomatic COVID-19, but acknowledges that individual clinical scenarios may lead clinicians toward testing and consideration of treatment. Individuals who have clinical or epidemiologic reasons that might make testing desirable (e.g., high-risk individuals, such as those who have pulmonary conditions or are immunocompromised or those in close contact with immunocompromised individuals) may be considered for testing. Testing should be done at least 5 days after the exposure. If symptoms develop before 5 days, the exposed individual should get tested immediately[3]. Knowledge that an individual is infected with SARS-CoV-2 can be helpful to inform appropriate isolation. The decision to test asymptomatic persons should depend on the availability of testing resources. Known exposures are defined herein as close contact for at least 15 minutes over a 24-hour period with someone who has laboratory-confirmed COVID-19. Suspected exposures might be defined as working or residing in a congregate setting (e.g., long-term care or correctional facility, cruise ship, factory) experiencing a COVID-19 outbreak. The risk of contracting SARS-CoV-2 may vary under different exposure conditions, e.g., length of time exposed, indoor versus outdoor setting, whether masks were routinely worn. Household contacts may be especially high-risk. This recommendation assumes the exposed individual was not wearing appropriate PPE.
Summary of the evidence
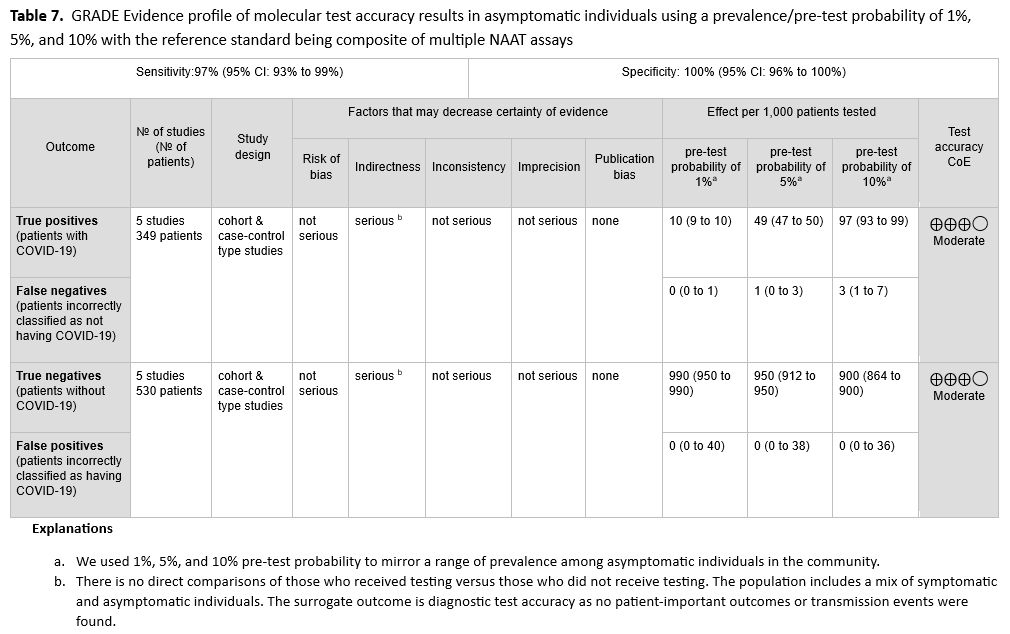
No direct evidence that assessed patient- or population-centered outcomes of testing versus no testing in asymptomatic individuals was found. Therefore, the panel relied on diagnostic test accuracy data to inform this recommendation. The reference standard was a composite of more than two standard NAATs (i.e., rapid RT-PCR and laboratory-based NAAT). Five studies [21-25] with 349 positive and 530 negative individuals, based on standard NAAT, were used to inform this recommendation. The pooled sensitivity was 97% (95% CI: 93 to 99) and the pooled specificity was 100% (95% CI: 96 to 100). (See Figures s2a-s2b). We considered 1%, 5%, and 10% as prevalences of COVID-19 in asymptomatic individuals with known exposures. Across this range of prevalences, there were no predicted false-positive and 0 to 3 predicted false-negative results per 1,000 individuals. (See Table 7). The certainty of the evidence was moderate due to down rating for indirectness. The indirectness was due to lack of direct comparison of those who received testing versus those who did not receive testing. Also, the population included a mix of symptomatic and asymptomatic persons.
Benefits and harms
Testing asymptomatic individuals who have been exposed, or suspected to have been exposed, to SARS-CoV-2, allows for isolation of those who are positive. Whether in an institutional cluster or a wider community outbreak, isolation has the potential to help reduce SARS-COV-2 transmission. A positive result will reinforce the importance of isolation as well as inform contact tracing, cohorting, or other mitigation strategies. There is potential harm in a false negative NAAT result collected from an exposed individual who is infected; these individuals may incorrectly consider themselves non-infected, and unknowingly expose others to SARS-CoV-2 as a result. Some individuals who test negative may be in the incubation phase of disease when tested. They may subsequently develop active infection and viral shedding, but incorrectly consider themselves uninfected.
Additional considerations
There are costs related to testing asymptomatic exposed individuals; since quarantine may still be indicated regardless of test results, such testing may add cost without changing practice. Data are limited to define definitions of close contact or high-risk exposure. Considerations when assessing the risk of a known contact may include the duration of exposure and certain clinical symptoms (e.g., cough) of the index case. Risk stratification of a given exposure can be made in consultation with public health authorities. The ideal time to test an asymptomatic contact of a known or suspected COVID-19 case is unknown. Timing of when to test or possible repeat testing also become complicated for household contacts with ongoing exposure.
Conclusions and research needs for this recommendation
Testing in asymptomatic subjects with known or suspected exposures should be coordinated with local public health officials. This indication for testing is especially important in situations where knowledge of asymptomatic or pre-symptomatic infection is essential for determining medical follow-up, defining risks for other vulnerable individuals in a household, congregate setting or hospital. Special consideration should also be given to healthcare personnel exposed without appropriate PPE in healthcare settings.
Comparative studies (preferably randomized controlled trials) along with cost-effectiveness analyses of testing strategies in asymptomatic populations are needed. Studies on the ideal time and collection method for testing asymptomatic individuals who have been exposed to COVID-19 should be performed. In addition, what constitutes an exposure that would justify testing requires further research. Whether early diagnosis of COVID-19 might provide an opportunity to intervene therapeutically and change the ultimate course of infection (e.g., prevent severe pneumonia) is unknown. If this is shown to be the case, the opportunity for therapeutic intervention might justify screening exposed individuals. The benefit might vary depending on underlying diseases, immune status, age, and vaccination status of the exposed individual, as well as the particular viral variant to which they were exposed and their prior history of COVID-19 infection (which might be unknown). Finally, recommendations for testing of asymptomatic individuals will likely change over time as the numbers and types of COVID-19 cases change.
Recommendation 7: Rapid versus Laboratory-based NAATs in Asymptomatic Individuals
Recommendation 7: For individuals who have clinical or epidemiologic reasons that might make testing desirable, the IDSA panel suggests using either rapid or laboratory-based NAATs in asymptomatic individuals with known exposure to SARS-CoV-2 infection (conditional recommendation, moderate certainty of evidence).
- Remarks:
- Appropriate specimen collection and transport to the laboratory or testing site are critical to ensure quality results; resources are available on the IDSA website. Definitions of rapid NAATs have varied; some, including the U.S. FDA, consider turnaround times less than or equal to 30 minutes to define rapid NAATs, whereas others use less than or equal to 60-minutes or even longer. This time is for testing only (inclusive of nucleic acid extraction) and does not include time between specimen collection and testing or time between testing and reporting. Rapid tests typically have few operator steps and may be amendable to testing near patients or even at the point-of-care performed by non-laboratory staff. Rapid test methodologies include rapid RT-PCR and rapid isothermal NAAT. Standard tests require instrumentation and/or processing that must typically be performed in a clinical laboratory by trained laboratory staff.
- This recommendation applies only to tests evaluated in the included studies. Variability of test performance with different specimen types may be important. The evaluated assays used diverse technologies (e.g., isothermal and non-isothermal test amplification) that may theoretically impact results. Limited data were available regarding the analytical performance of NAATs in immunocompromised or vaccinated individuals, in those who have had prior SARS-CoV-2 infection, or in those infected with different SARS-CoV-2 variants.
Summary of the evidence
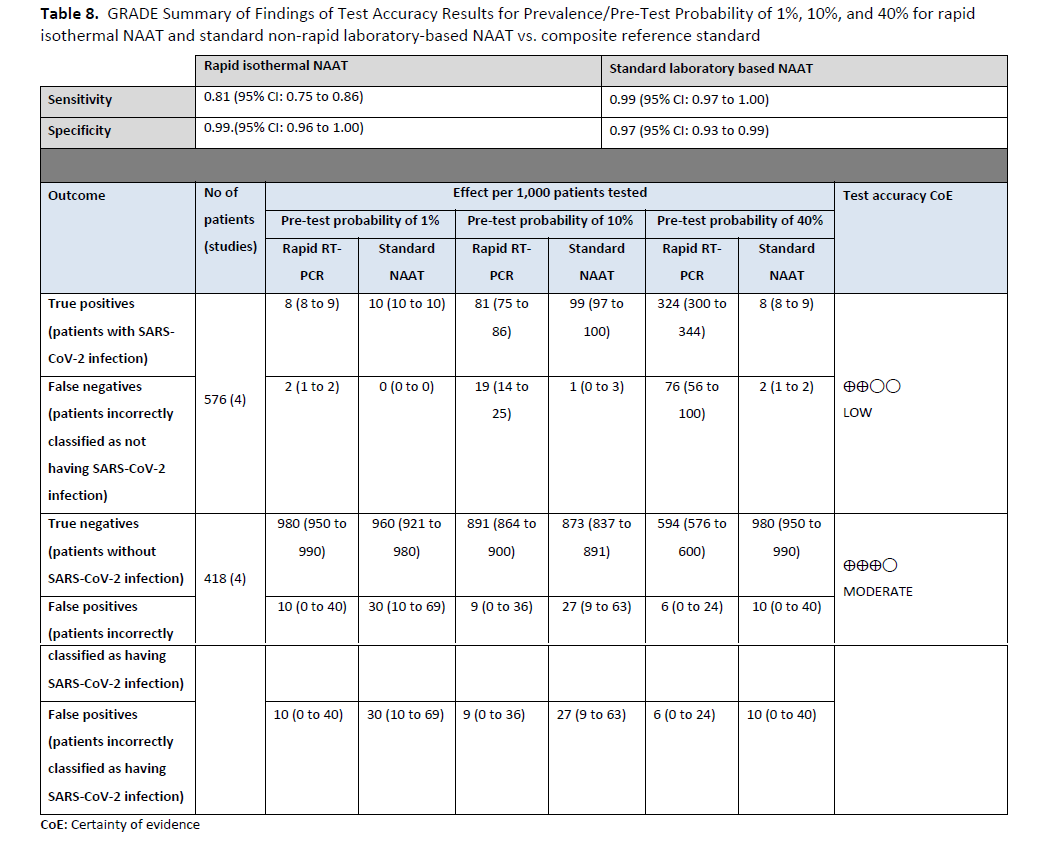
Three studies [85, 90, 91] with 181 positive and 3,088 negative individuals that used rapid and laboratory-based NAAT were identified to inform this recommendation (Figures s16a - s16b). The reference test used for this analysis was standard laboratory-based NAAT. The pooled sensitivity of rapid NAAT was 88% (95% CI: 81 to 92) and the pooled specificity was 99% (95% CI: 95 to 100). The IDSA panel considered 1%, 5%, and 10% as prevalences of SARS-CoV-2 infection in asymptomatic individuals with known SARS-CoV-2 exposure. Across these prevalences, the false-negative numbers ranged from 1 to 12 and the false-positive numbers from 9 to 10 per 1,000 persons tested. The certainty of the evidence was low due to concerns related to unexplained inconsistency in addition to the risk of bias with the use of laboratory-based NAAT as the reference standard. (See Table 8)
Benefits and harms
The value of rapid testing, ideally completed while the patient is physically present, is that it allows decisions about treatment, isolation, and contact tracing to be made quickly. Rapid testing may reduce patient anxiety while test results are pending. A rapid result decreases concerns about losing patients to follow up and generally simplifies follow up.
The important finding here is that test turnaround time is not intrinsically linked to poor diagnostic test accuracy. All rapid NAATs had very high specificities, minimizing false positive results. The potential harm of false positive results includes isolating individuals who do not have SARS-CoV-2 infection, which may cause unnecessary anxiety, as well as potentially administering unneeded COVID-19 therapies, increasing days away from work or school, and wasted time and energy directed at contact tracing. Positive results from rapid NAATs do not need to be confirmed routinely by standard laboratory based NAATs. In addition, the pooled sensitivity of the rapid tests of 88% implies that rapid NAATs, especially RT-PCR tests, perform just as well as non-rapid NAATs, meaning that negative rapid NAAT results do not need to be confirmed routinely by standard laboratory-based NAATs.
Additional considerations
There were limited data on the performance of rapid NAATs in children. In addition, the specific test, anatomic site of sampling, use or non-use of viral transport media and quality of specimen collection may impact test performance, regardless of whether a test is rapid or standard. Lastly, there were no studies directly comparing rapid isothermal NAAT and rapid RT-PCR tests to one another.
Conclusions and research needs for this recommendation
The specificity of rapid and standard laboratory based NAATs is equivalent. The sensitivity of rapid NAATs, especially RT-PCR and standard laboratory-based NAAT are similar. Rapid NAATs for SARS-CoV-2 and other microorganisms are an important advance in healthcare and open the door to new ways of healthcare delivery.
Recommendation 8: NAAT in Asymptomatic Individuals before Hospital Admission
Recommendation 8: The IDSA panel suggests against routine SARS-CoV-2 NAAT in asymptomatic individuals without a known exposure to COVID-19 who are being hospitalized (conditional recommendation, very low certainty evidence).
- Remarks:
- Important considerations for this recommendation are that the IDSA panel was unable to identify studies published during the period of literature review that showed reduced SARS-CoV-2 transmission to healthcare providers or to other patients resulting from prehospitalization testing. The evidence was indirect and assessed only diagnostic test accuracy in studies of symptomatic patients alone or together with asymptomatic patients. The burden of testing all patients planned to be admitted was considered, in the face of limited evidence. Finally, there are other effective infection prevention interventions, including use of PPE and vaccination that should be considered.
- The panel acknowledges that there could be a benefit of pre-admission NAAT in some situations, such as admission to a multibed room; to a unit with a congregate treatment area, such as a behavioral health unit; or to a positive pressure room or unit .
Summary of the evidence
We did not identify any studies that directly assessed a strategy of nucleic acid testing for SARS-CoV-2 versus no testing before hospitalization for patients who were admitted for diagnoses unrelated to COVID-19. We also did not identify test accuracy studies assessing the performance of SARS-CoV-2 viral RNA tests in asymptomatic individuals as compared to a composite reference standard. For that reason, we applied the test accuracy reported from studies of symptomatic and mixed (symptomatic and asymptomatic, or symptom status not specified) patient populations to asymptomatic populations of patients who were being admitted to hospital with pre-test probabilities of 1%, 5%, and 10%. The reference pooled sensitivity used was 97% (95% CI: 93% to 99%) and the reference pooled specificity used was 100% (95% CI: 96% to 100%). (See Figures s2a-s2b). However, these test accuracy values, particularly the value for sensitivity, may be higher than the true accuracy of testing in asymptomatic individuals, therefore representing a best-case scenario. If instead, the reference used was derived from three cohort studies [92-94] that performed head-to-head comparisons of two NAATs, the sensitivity ranged between 85% and 93% when comparing a single NAAT to any positive test. The significant limitations associated with the available evidence led to very low certainty of the effect of testing overall based on very serious indirectness and serious inconsistency.
Benefits and harms
Although isolation and cohorting of patients with asymptomatic SARS-CoV-2 infection are interventions that may reduce healthcare-associated SARS-CoV-2 transmission, there is no direct evidence to suggest that routine screening of asymptomatic individuals prior to admission confers significant benefit. In contrast, there is significant burden on the institution both logistically and financially, and also potential harm to patients in delaying admission. Benefit may be higher in healthcare facilities with many multibed rooms, in behavioral health settings, on positive pressure wards such as hematopoietic cell transplantation wards, or in situations where PPE is limited, especially when community transmission levels are moderate or high.
Additional considerations
Given evidence that wearing masks and other PPE can prevent SARS-CoV-2 transmission and factoring in other considerations such as availability of private patient rooms and vaccination status of patients and healthcare providers, routine screening of asymptomatic individuals before hospital admission may not result in added benefit.
Conclusions and research needs for this recommendation
Routine pre-hospitalization screening of asymptomatic individuals may not provide added benefit to mitigation steps already in place, such as universal masking by healthcare providers. Studies specifically addressing possible benefits of routine pre-hospitalization screening of asymptomatic patients by NAAT in various settings is needed.
Recommendation 9: NAAT in Asymptomatic Individuals Undergoing Procedures
Recommendation 9: The IDSA panel suggests against routine SARS-CoV-2 NAAT of asymptomatic individuals without a known exposure to COVID-19 who are undergoing a medical or surgical procedure (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on non-standardized instrument signal values, such as cycle threshold (Ct) values.
- Decisions on the timing of a procedure in a patient with prior SARS CoV-2 infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age), procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
- Given limited evidence for poor outcomes in asymptomatic persons who undergo major surgery soon after testing positive for SARS-CoV-2 infection, testing may be considered during periods of high community transmission.
- Testing may also be considered before solid organ transplantation, hematopoietic stem cell transplantation or CAR-T cell therapy.
- This recommendation applies to settings where protective measures, such as PPE, are available and are used with adherence. Other factors to consider include the vaccination status of healthcare providers and patients, and whether patients will be roomed with other patients before or after the procedure. This recommendation is based on general exposure in the community as compared to a specific known exposure.
Summary of the evidence
We did not identify any studies that directly assessed a strategy of testing asymptomatic individuals for SARS-CoV-2 infection versus no testing before a medical procedure with an outcome of reduced SARS-CoV-2 transmission. We also did not identify test accuracy studies that directly assessed the performance of SARS-CoV-2 NAATs in asymptomatic individuals who were undergoing medical procedures.
We identified two studies [95, 96] that found almost no SARS-CoV-2 infection in healthcare providers or patients who had performed upper or lower endoscopies on asymptomatic patients who had not undergone pre-procedure SARS-CoV-2 NAAT. Both studies occurred before emergence of the Omicron variant. In the first study, [95] 29 staff members who worked in an endoscopy unit for at least 2 days a week for at least 6 months were followed for 20 months without any pre-procedural testing of patients, during which time 0/29 staff members contracted COVID-19. The vaccination rate of the team was 97%. In the second study, [96] , retrospective review of 214 endoscopy patients who were tested for SARS-CoV-2 after the procedure for unspecified indications identified only one who developed a positive SARS-CoV-2 NAAT 29 days post procedure.
We also identified six different studies [96-101] in which pre-procedural testing of asymptomatic patients was performed before an endoscopy procedure. In these studies, less than 1% of patients tested positive for SARS-CoV-2 before their procedure, and subsequently had their procedures delayed. Also in those studies, 0-4% of healthcare providers who cared for the patients were reported to have positive SARS-CoV-2 test results sometime after the procedure. These studies spanned different waves of the pandemic, reported differing availability of personal protection equipment, and varying vaccination status.
Due to the concerns with the significant limitation of the available evidence, the panel agreed that the overall certainty of the evidence supporting this recommendation was very low.
Benefits and harms
The possible benefit of screening asymptomatic individuals include reducing exposure of healthcare providers and other patients to SARS-CoV-2, thereby reducing SARS-CoV-2 transmission. Potential harms include unnecessary delays in procedures for patients, and false confidence among healthcare providers who are exposed to a patient who is in the incubation period for COVID-19 when tested before the procedure but who would test positive and be infectious later during the procedure. Institutions that require pre-procedure screening generally request testing within 72 hours of the scheduled procedure. Requiring a shorter screening window might result in more accurate prediction of infection risk at the time of the procedure but may be unrealistic for most institutions, especially if testing is performed in an off-site laboratory. Wider use of point-of-care NAAT may also be difficult in many settings given testing logistics, supply chain concerns, and limited tests with an indication for use in asymptomatic individuals.
In cases of positive results, depending on the procedure, deferring when there are other risk mitigation measures in place, including vaccination of healthcare providers and use of appropriate PPE, may cause undue harm to the patient and have minimal or no effect on risk of transmission from the index patient. This may be especially true for patients who have recovered from COVID-19 and are no longer infectious, but who continue to shed non-viable SARS-CoV-2 that is detected by highly sensitive NAATs. The panel placed special emphasis on the logistics of re-scheduling procedures and increased risk associated with delaying procedures. Even in cases where a healthcare provider or another patient contracts COVID-19, linking a SARS-CoV-2 acquisition to an asymptomatic individual is difficult, especially when community transmission levels are high.
Additional considerations
The panel recognized that different procedures may carry different transmission risks. While there is a lack of direct evidence, a systematic review of literature from 1990 to 2010 on SARS transmission showed that some aerosol generating procedures carried increased risk of transmission to healthcare providers. Specifically, the investigators found that tracheal intubation carried the most risk, followed by non-invasive ventilation, tracheotomy, and manual ventilation. In contrast, the investigators found no significant increase in risk during other intubations, endotracheal suction, bronchoscopy, suction of body fluids, nebulizer treatment, administration, and manipulation of supplemental oxygen mask or BiBAP mask, defibrillation, chest compression, insertion of nasogastric tube, or collection of expectorated sputum. The investigators emphasized that use of appropriate PPE is an important mitigating factor, and that in their analysis, it was not possible to assess and exclude transmission events due to non-compliant or inappropriate use of PPE.
Despite limited evidence, testing asymptomatic individuals before solid organ transplantation or cellular therapy (hematopoietic cell transplantation or CAR-T cell therapy) is recommended at this time by several professional medical societies[102].
Conclusions and research needs for this recommendation
Routine pre-procedure screening of individuals with very low pre-test probability of SARS-CoV-2 infection may not provide benefit, based on the evidence identified by the panel. Factors such as vaccination status, availability of appropriate PPE, and risk assessment of procedures should be considered when considering pre-procedure screening. Studies comparing these risks and mitigating measures would be helpful in answering this question more definitively.
Recommendation 10: Repeat Testing in COVID-19 Patients Requiring Procedures
Recommendation 10: The IDSA panel suggests against routinely repeating NAAT before medical or surgical procedures in patients with a recent history of COVID-19 (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens without evidence of infectious virus has been reported widely.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
- Decisions on the timing of a procedure in a patient with prior SARS CoV-2 infection must balance the risk to the patient against the risks of delaying or avoiding the planned procedure, and should consider patient-related factors (e.g., vaccination status, symptomatic status, age), procedure-related factors (e.g., level of urgency, whether procedure generates aerosols), and procedural area infection control practices.
Summary of the evidence
We did not identify any studies that directly assessed a strategy of NAAT testing for SARS-CoV-2 versus no testing in patients with a prior diagnosis of COVID-19. The patient-centered outcomes of interest include risk of complications or poor procedural outcomes related to SARS CoV-2 infection, as well as risk of transmission to healthcare providers and other patients.
We identified five cohort studies [103-107] that addressed the prognosis for individuals with SARS-CoV-2 infection who underwent procedures. The surgeries included in those studies are all operative procedures that are routinely performed in an operating theater by a surgeon, which may vary in the level of surgical difficulty. Some studies excluded emergency surgery from the analysis [106, 107] while the rest included emergency and non-emergency operations [103-105]. In all studies, the outcomes were correlated to time since COVID-19 diagnosis. The diagnostic criteria for SARS-CoV-2 infection differed by study, ranging from NAAT to chest imaging plus symptoms. No repeat testing strategies were identified.
Data from early in the pandemic indicate that individuals with peri-operative SARS-C-V-2 NAAT positivity may have had an increased risk of post-operative complications [103, 104]. The symptom status of patients may have played a role in that symptomatic patients had a higher reported risk of complications [105]. Among patients with SARS-CoV-2 infection who undergo operative procedures, there may be an increased risk of complications if the procedure is performed within 0-8 weeks of diagnosis, but not after 8 weeks [105-107]. Vaccinated individuals may have a reduced risk of post-procedural complications. [106]. In contrast, a study published after the literature review for this guideline was completed but that included patients from early in the COVID-19 pandemic found a decreasing risk of major postoperative cardiovascular events that was associated with time from an initial positive diagnostic SARS-CoV-2 test result (adjusted OR [aOR], 0.99 [per 10 days after diagnosis of SARS-CoV-2 infection]; 95% CI, 0.98-1.00; P = .006.) [108] This association held for patients who had had at least one dose of SARS-CoV-2 vaccine.
None of these studies addressed whether results of repeat SARS-CoV-2 molecular testing could help to predict the risk of post-operative morbidity in patients with a recent history of SARS-CoV-2 infection. Due to significant limitations of the available evidence, the IDSA panel agreed that overall certainty of evidence was very low.
Benefits and harms
SARS-CoV-2 infection, especially early in the pandemic, was associated with adverse post-operative outcomes, including an increased risk of vascular thrombosis, prolonged intubation in patients who undergo thoracic surgery, and increased mortality [103, 109, 110]. The IDSA panel found no evidence that repeating NAAT in a patient with a recent diagnosis of SARS-CoV-2 infection would help to mitigate that risk. Potential harms of testing include unnecessarily delaying a procedure in a patient because of a positive test, or risk of infection transmission to healthcare providers or other patients if appropriate transmission-based precautions are not used when caring for a patient with a negative test but who is still infectious.
Additional considerations
None.
Conclusions and research needs for this recommendation
The existing evidence surrounding postoperative complications in patients with recent SARS-CoV-2 infection is derived from time-based studies, with heterogeneous definitions of infection. Given the known prolonged positivity of molecular assays such as NAAT, further research is needed to correlate molecular test results and patient outcomes.
Recommendation 11: NAAT to Remove Patients with SARS-CoV-2 Infection from Isolation
Recommendation 11: The IDSA panel suggests against routinely repeating NAAT in patients with COVID-19 to guide release from isolation (conditional recommendation, very low certainty evidence).
- Remarks:
- NAAT is used to determine presence of SARS-CoV-2 RNA, which may not represent infectious virus.
- Detection of SARS-CoV-2 RNA in respiratory specimens for prolonged periods without evidence of infectious virus has been reported widely. Predicating release from isolation on a negative SARS-CoV-2 NAAT may extend the duration of isolation unnecessarily.
- Conversely, the IDSA panel was unable to find definitive evidence demonstrating that a negative NAAT result following a positive result is proof that a patient is no longer infectious.
- The IDSA panel concluded that data were insufficient to establish SARS-CoV-2 infectiousness of a patient based on Ct value results.
Summary of evidence
We identified no studies that directly assessed a strategy of testing versus not testing for SARS-CoV-2 to release patients from isolation. No studies included key patient outcomes such as transmission events, removal of isolation or return to work.
We identified four cohort studies [111-114] that assessed the course of positive SARS-CoV-2 NAAT results in the pre-Omicron era by serially testing patients. In a study of American National Football League players, among those who initially tested positive by NAAT, 146 (84%) tested negative before day 10 (defined as either a negative result or Ct value >35) [111]. In a study evaluating household transmission, over 50% of tested individuals undergoing serial NAAT continued to test positive on day 20 [112]. In another study, 61 of 207 (29%) outpatients and inpatients in a COVID Care Center remained NAAT positive until days 15-30 [113]. A separate study involving household and non-household exposed contacts noted a mean duration of positive RNA molecular assays of 14.6 days [114]. The duration of positive NAAT was reported to be consistently longer than the duration of a positive viral culture in two studies [112, 114]. All studies were heterogenous in terms of study population, different waves of the pandemic, variants and vaccination status of participants. Due to the significant limitation of the available evidence, the IDSA panel agreed that overall certainty of evidence was very low.
Rheumatologic disease review
The prevalence of SARS-CoV-2 infection in the seven studies of patients with rheumatologic disease ranged from 0.2 to 47.2% (median 0.8%). The rate of hospitalization ranged from 58.5-70.0% (median 68.8%, four studies), with an intensive care admission rate of 3.4-9.8% (median 5.9%, three studies), and a death rate of 0.0-26.3% (median 9.8%, seven studies). We identified three retrospective cohort studies that compared outcomes of COVID-19 in patients with and without rheumatologic diseases, and in patients on and off treatment for rheumatologic diseases [150, 160, 161]. Overall, there was no association between the presence of rheumatologic diseases, or their treatments, and poor outcomes in patients with COVID-19.
Benefits and harms
Potential benefits of NAAT to remove a patient from isolation might be definitive proof that a patient is not infectious. However, such proof does not exist. Potential harms of using NAAT to remove a patient from isolation include unnecessary prolongation of isolation and its attendant consequences (e.g., unnecessary absence from work, school, or recreational activities). Unnecessary financial costs would also accrue and should be considered.
Additional considerations
The decision to remove an individual from isolation involves assessment of the risk of that person’s contagiousness. While replication-competent virus as determined by viral culture has sometimes been used as a surrogate for infectious virus, viral culture is known to be less sensitive for diagnosis of SARS-CoV-2 infection compared to NAAT, and viral culture methods for SARS-CoV-2 are not standardized. NAAT testing remains positive for days to weeks longer than viral culture. Many factors in addition to the presence of replication-competent virus can affect transmission risk, including immune status and vaccination history of the index patient and their contacts and whether unidirectional or bidirectional masks are worn; length of time of the exposure and ventilation of the space in which exposure occurs. The presence of replication-competent virus in the setting of an outdoor walk between two masked individuals will be associated with a much lower risk of infection than if the two individuals meet in a poorly ventilated room without masks. Additionally, while it seems intuitive that a negative NAAT result following a positive result should correlate with viral clearance and lack of ongoing infectivity, this “test of cure” concept has not been confirmed.
Conclusions and research needs for this recommendation
The lack of a gold standard of infectiousness is a major hindrance in the evaluation of any diagnostic test methodology which is used to detect infectiousness. Development of a laboratory surrogate of contagiousness and of assays for test-of-cure (i.e., test of non-infectiousness) would be of value.
Recommendation 12: Molecular Testing at Home
Recommendation 12: The IDSA panel suggests neither for nor against home-testing for SARS-CoV-2. (evidence gap).
- Remarks:
- The panel defined time-sensitive surgery as medically necessary surgeries that need to be done within three months.
- Testing should ideally be performed as close to the planned surgery as possible (e.g., within 48-72 hours).
- To limit potential poor outcomes, deferring non-emergent surgeries should be considered for patients testing positive for SARS-CoV-2.
- Decisions about PPE use for the aerosol generating portions of these procedures may be dependent on test results when there is limited availability of PPE. However, there is a risk for false negative test results, so caution should be exercised by those who will be in close contact with/exposed to the upper respiratory tract (e.g., anesthesia personnel, ENT procedures).
- The decision to test asymptomatic patients will be dependent on the availability of testing resources.
- This recommendation does not address the need for repeat testing if patients are required to undergo multiple surgeries over time.
Summary of the evidence
We identified no studies that directly compared a strategy of home testing versus standard laboratory-based NAAT for SARS-CoV-2 to determine test accuracy. “Home testing” was defined as the study authors stating that a test was performed by a patient in their home. We identified only one study [64] that assessed home testing using a SARS-CoV-2 NAAT. The reported outcome was diagnostic test accuracy. In this study, a NP swab was collected by a healthcare provider in an outpatient healthcare facility and tested. The patient collected a second MT swab specimen while at home within 1 day of the healthcare provider specimen collection and testing. Home testing had a sensitivity of 28/35 (80.0%) and specificity of 140/143 (97.9%) when the health care worker NP swab test result was used as the reference test. The lack of consideration of other factors (e.g., sample site), confound the ability to assess the effect of home testing alone.
Benefits and harms
The certainty of evidence supporting home NAAT for SARS-CoV-2 is too limited to draw conclusions about benefits or harms of this approach. Key concerns relate to whether sampling at home would lead to lower sensitivity of testing.
Additional considerations
Although over-the-counter home molecular tests are available, they are generally more expensive on a per test basis than antigen tests; some home molecular tests require purchase of a reader at an additional cost [9]. These operational challenges and increased costs bring into question the benefits of home molecular testing, especially when compared with the ease and lower cost of home antigen testing.
Conclusions and research needs for this recommendation
If the desire for home SARS-CoV-2 molecular tests rises (e.g., if a new viral variant were identified necessitating widespread home sampling), and the logistical challenges of specimen type, rapid transport and/or in-home testing and resulting could be resolved, research would need to carefully assess the accuracy of this approach versus specimen collection and testing in a healthcare setting.
Discussion
Molecular tests designed to detect SARS-CoV-2 nucleic acids have become the gold standard technology for confirming a diagnosis of COVID-19. Although the U.S. was hampered by limited test availability early in the outbreak, there are now more than 275 different commercially available molecular SARS-CoV-2 assays, including laboratory-based, rapid, point-of-care, and over-the-counter home tests, with SARS-CoV-2 NAATs now available in most areas of the US. Most tests were granted emergency use authorization by the US FDA; to date, only one molecular assay has been granted a de novo, 510(k) clearance [115], which is the usual pathway for FDA approval of an in vitro medical device. Despite their EAU status, SARS-CoV-2 NAATs have demonstrated high levels of accuracy and reproducibility, allowing diagnostic testing to play an essential role in the clinical management of patients with COVID-19 and in guiding public health responses aimed at curbing the pandemic. While the Department of Health and Human Services is planning for the federal COVID-19 Public Health Emergency to expire on May 11, 2023 [116], there has been no similar declaration to terminate the independent EUA for SARS-CoV-2 diagnostic tests, and it is expected that the EUA will remain in effect after this date [117]. If the EUA declaration is terminated, advance notice will be published in the federal register, which will start the transition process to normal operations.
The approval of SARS-CoV-2 diagnostic tests through EUA may have impacted recommendations for diagnostic testing indirectly. EUA requires submission to FDA of analytical performance data but not clinical performance data. Perhaps because of these requirements, the IDSA panel identified few studies that reported clinical performance of a test; most recommendations in this guideline are based instead on diagnostic test accuracy. Metrics to measure diagnostic test accuracy include sensitivity, which is the ability of the test to correctly identify those with infection, and specificity, the ability of the test to correctly identify those without the disease. The positive and negative predictive values of the test are also essential for interpreting test results. Calculation of predictive values requires information about the prevalence or pre-test probability of SARS-CoV-2 infection. In practice, however, the true prevalence of COVID-19 in the community may not be well-defined and may be underestimated when test availability is limited or when test results are not reported to public health authorities. Recognizing these complexities, the IDSA panel varied estimates of prevalence/pre-test probability and assay sensitivity and specificity in our analyses based on the available literature and public dashboards to mirror what may be encountered in clinical practice.
The number of published, peer-reviewed studies that informed the current and third iteration of the IDSA guideline on molecular diagnostic testing for SARS-CoV-2 is substantially increased compared to the number available to inform the second version, which was published in 2021. The larger number of articles allowed the panel to exclude from the literature review preprints that had not undergone peer review and resulted in greater certainty of evidence to support some recommendations. For example, the level of certainty of evidence to support a recommendation of NAAT for symptomatic individuals suspected of having COVID-19 was deemed very low in 2021 and moderate today. Still, most recommendations in this guideline are conditional, with very low or low certainty of evidence to support them. Evidence was especially sparse or of poor quality for performance of tests in children, immunocompromised individuals, those who had been vaccinated or infected previously with SARS-CoV-2, and persons infected with newer SARS-CoV-2 variants such as Omicron. Other research needs recognized by the panel include identification of a laboratory marker of infectiousness that could be used to guide release of patients with symptomatic or asymptomatic COVID-19 from isolation, and studies on the value of testing for asymptomatic SARS-CoV-2 infection at the time of hospitalization or before a medical or surgical procedure. Additional data on the accuracy of home testing, and on testing specimen sources other than NP swabs, are also needed. Ideally, clinical test performance should be determined in prospective multicenter studies using a well-defined reference standard as the benchmark for test comparisons. Table 9 outlines the type of clinical studies needed to address the most pressing COVID-19 diagnostic knowledge gaps.
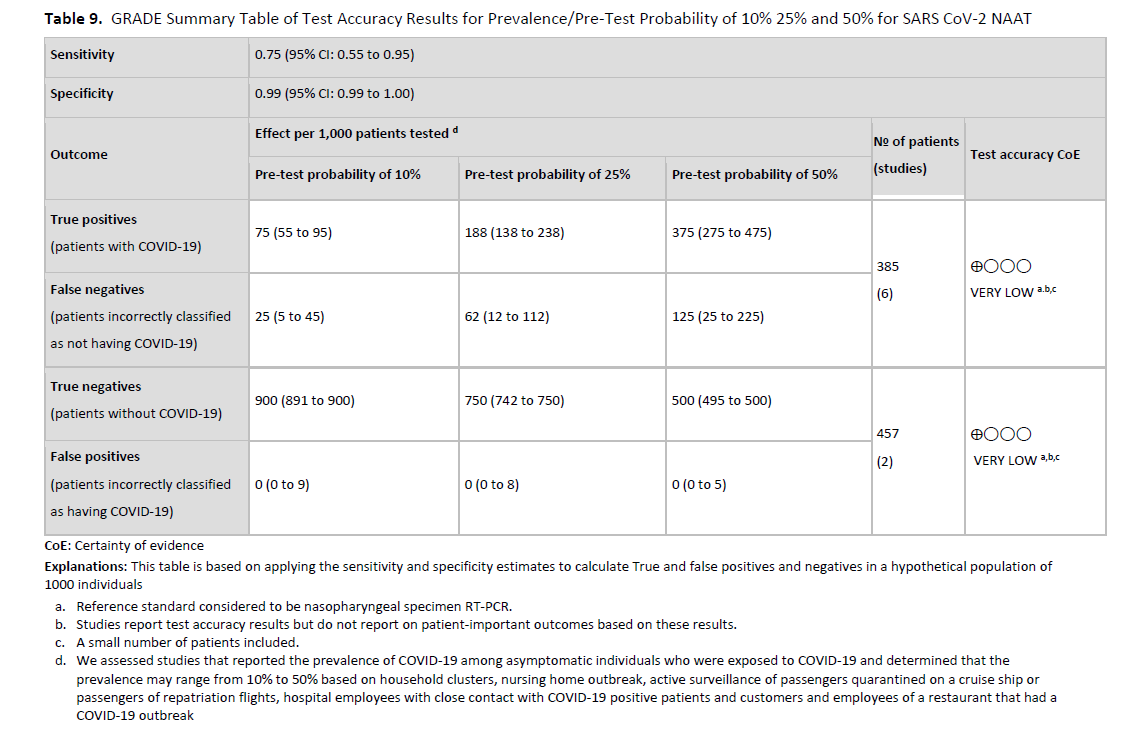
Conclusions
The guideline panel used a methodologically rigorous process to critically appraise the available diagnostic literature and update SARS-CoV-2 molecular testing recommendations. The evidence available for most recommendations remained sparse or of low quality. Based on moderate certainty evidence, the IDSA panel recommends NAAT for all symptomatic individuals suspected of having COVID-19. The IDSA panel also suggests testing a specimen collected by a healthcare provider or patient from the nasopharynx or alternative upper respiratory tract specimens once rather than repeat testing. In addition, testing selected asymptomatic individuals is suggested when the results will have significant impact on isolation/quarantine/usage of PPE. The IDSA panel suggests against routine NAAT of asymptomatic individuals before hospitalization or a medical/surgical procedure, although the panel recognizes that there may be benefit to testing when PPE is limited, patients must be housed in semi-private or multibed rooms, in behavioral health environments, before solid organ or hematopoietic stem cell transplantation, or when community transmission levels are high. The panel suggests against repeat NAAT to guide removal of isolation in patients with COVID-19, given known prolonged viral RNA shedding that may not represent live virus, and the lack of documentation that a negative NAAT always correlates with zero risk of transmission. An evidence gap precluded making a recommendation about home testing. The critical components of future COVID-19 diagnostic studies include use of a well-defined reference standard with detailed descriptions of specimen types, collection methods and their timeframe after symptom onset or exposure to a laboratory-confirmed case.
Notes
Acknowledgement
The expert panel thanks the Infectious Diseases Society of America for supporting guideline development, and specifically the Executive Committee of the IDSA Board of Directors as well as IDSA staff members Dana Wollins, Genet Demisashi, Sheila Tynes, Hannah Rehm, and Imani Amponsah for their continued support throughout the guideline process. The panel also expresses its appreciation to the members of SHEA, PIDS, and ASM who provided their thoughtful and comprehensive review.
Financial Support
This project was funded in part by a cooperative agreement with the Centers for Disease Control and Prevention (CDC) (grant number 6 NU50CK000477-04-01). The CDC is an agency within the Department of Health and Human Services (HHS). The contents of this guideline do not necessarily represent the policy of CDC or HHS and should not be considered an endorsement by the Federal Government.
COI Summary
The following list displays what has been reported to the IDSA. To provide thorough transparency, the IDSA requires full disclosure of all relationships, regardless of relevancy to the guideline topic. Evaluation of such relationships as potential conflicts of interest is determined by a review process which includes assessment by the Board of Directors liaison to the Standards and Practice Guideline Committee and, if necessary, the Conflicts of Interest (COI) and Ethics Committee. The assessment of disclosed relationships for possible COI is based on the relative weight of the financial relationship (i.e., monetary amount) and the relevance of the relationship (i.e., the degree to which an association might reasonably be interpreted by an independent observer as related to the topic or recommendation of consideration). The reader of these guidelines should be mindful of this when the list of disclosures is reviewed. M.H. serves on a clinical adjudication panel for Sanofi; receives research funding from the Centers for Disease Control and Prevention (CDC) and CDC Foundation; serves on the Society for Healthcare Epidemiology of America (SHEA) Board of Directors and Chair of the SHEA Education & Research Foundation; received other numerations from Sage, Medline, and Molnylycke; and served as Chair of the IDSA Diagnostics Committee. K.H. served an advisor to Quidel, BioFire, Pfizer, and Takeda; received other numerations from Quidel, Pfizer and Takeda; served Editor to American Society of Microbiology (ASM) and member of Clinical and Laboratory Standards Institute Antifungal Committee; received research funding from the National Institutes of Health (NIH); and served on the exam committee for the American Board of Internal Medicine, and associate editor for Open Forum Infectious Diseases. J.E. serves as a consultant for Sanofi Pasteur, Pfizer, and AstraZeneca; an advisor/consultant for Meissa Vaccines; receives research funding from the CDC, Pfizer, Brotman Baty Research Institute, Merck, Novavax, GlaxoSmithKline, and AstraZeneca; served as an advisor to Teva Pharmaceuticals; and served as member of Pediatric Infectious Diseases Society (PIDS) Publication Committee and Transplant ID Committee. M.L. serves as an advisor for Sanofi, Seqirus, Medicago, GSK, Janssen, Novavax, Pfizer, MD Brief; receives research funding from the Canadian Institutes of Health Research, World Health Organization (WHO), Medical Research Council (United Kingdom), has received in-kind supply of vaccine from Sanofi, has been paid for expert testimony on institutional and workplace vaccine policy, and has served on the DSMB for CanSino Biologics and an advisor to Merck. R.P has a patent on Bordetella pertussis/parapertussis PCR issued, a patent on a device/method for sonication with royalties paid by Samsung to Mayo Clinic, and a patent on an anti-biofilm substance issued; serves as consultant to PhAST, Torus Biosystems, Day Zero Diagnostics, Mammoth Biosciences, Netflix, Abbott Laboratories, Oxford Nanopore Technologies, CARB-X, Qvella, and HealthTrackRx; receives other numeration from NBME, UpToDate, and the Infectious Disease Board Review Course; received grants from CD Diagnostics, Merck, Hutchison Biofilm Medical Solutions, Accelerate, ContraFect, TenNor Therapeutics Limited, Shionogi, NIH, BIOFIRE, Adaptive Phage Therapeutics, National Science Foundation, and the Department of Defense; and has served as a consultant to Curetis, Specific Technologies, NextGen Diagnostics, Pathoquest, Selux Diagnositcs, and 1928 Diagnostics. S.S serves as a Board member for the Evidence Foundation, receives honoraria for evidence reviews, methodological support and teaching from the Evidence Foundation; serves on guideline panels for the American Gastroenterological Association (AGA); and receives research funding from the Department of Veterans Affairs Evidence Synthesis Program. Y.F.Y. serves as a Board member for the Evidence Foundation; receives honoraria for evidence reviews, methodological support and teaching from the Evidence Foundation, the AGA for evidence reviews, and the Institute for Clinical and Economic Review (ICER) for committee meetings; serves as a Director for the Evidence Foundation and for the U.S. GRADE Network; and served on an Independent Appraisal Committee for ICER. R.M serves as a Board member for the Evidence Foundation; and receives honoraria for evidence reviews, methodological support and teaching from the Evidence Foundation. M.H.M serves as a Board member for the Evidence Foundation; receives honoraria for evidence reviews, methodological support and teaching from the Evidence Foundation; receives research funding from the Agency for Healthcare Research and Quality, the Endocrine Society, and the Society for Vascular Surgery; has received research funding the American Society of Hematology and the WHO; and has served as a guideline methodologist for the WHO. A.B. received honorarium from the ICER. R.A.M serves as a Board member for the Evidence Foundation; receives honoraria for evidence reviews, methodological support and teaching from the Evidence Foundation, and ICER for committee meetings; receives research funding from the NIH, the WHO, the American College of Rheumatology, the American Society of Hematology, and Bohringer Ingelheim; serves as Chair of the Midwest Comparative Effectiveness Public Advisory Council of the ICER; serves on the Methods Committee for Kidney Disease Improving Global Outcomes Work Group; serves on the Clinical Guidelines Committee for the Canadian Society of Nephrology; and previously served on the Clinical Guidelines Committee for the American College of Physicians (ACP). All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed. All other authors report no potential conflicts.
IDSA Disclaimer
It is important to realize that guidelines cannot always account for individual variation among patients. They are assessments of current scientific and clinical information provided as an educational service; are not continually updated and may not reflect the most recent evidence (new evidence may emerge between the time information is developed and when it is published or read); should not be considered inclusive of all proper treatments, methods of care, or as a statement of the standard of care; do not mandate any particular course of medical care; and are not intended to supplant physician judgment with respect to particular patients or special clinical situations. Whether and the extent to which to follow guidelines is voluntary, with the ultimate determination regarding their application to be made by the physician in the light of each patient’s individual circumstances. While IDSA makes every effort to present accurate, complete, and reliable information, these guidelines are presented “as is” without any warranty, either express or implied. IDSA (and its officers, directors, members, employees, and agents) assume no responsibility for any loss, damage, or claim with respect to any liabilities, including direct, special, indirect, or consequential damages, incurred in connection with these guidelines or reliance on the information presented.
The guidelines represent the proprietary and copyrighted property of IDSA. Copyright 2023 Infectious Diseases Society of America. All rights reserved. No part of these guidelines may be reproduced, distributed, or transmitted in any form or by any means, including photocopying, recording, or other electronic or mechanical methods, without the prior written permission of IDSA. Permission is granted to physicians and health care providers solely to copy and use the guidelines in their professional practices and clinical decision-making. No license or permission is granted to any person or entity, and prior written authorization by IDSA is required, to sell, distribute, or modify the guidelines, or to make derivative works of or incorporate the guidelines into any product, including but not limited to clinical decision support software or any other software product. Except for the permission granted above, any person or entity desiring to use the guidelines in any way must contact IDSA for approval in accordance with the terms and conditions of third-party use, in particular any use of the guidelines in any software product.
References
-
- Hanson KE, Caliendo AM, Arias CA, et al. The Infectious Diseases Society of America Guidelines on the Diagnosis of COVID-19: Molecular Diagnostic Testing. Clin Infect Dis 2021; ciab048: Available at: https://doi.org/10.1093/cid/ciab048 [Epub ahead of print 22 January 2021].
- Hayden MK, Hanson KE, Englund JA, et al. The Infectious Diseases Society of America guidelines on the diagnosis of COVID-19: antigen testing. Clinical Infectious Diseases 2023: ciad032.
- Centers for Disease Control and Prevention. Overview of Testing for SARS-CoV-2, the virus that causes COVID-19. Available at: https://www.cdc.gov/coronavirus/2019-ncov/hcp/testing-overview.html#:~:text=People%20who%20have%20had%20an,CDC's%20COVID%2D19%20isolation%20guidance. Accessed 15 June 2023.
- Zhu N, Zhang D, Wang W, et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 2020; 382(8): 727-33.
- Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020; 395(10224): 565-74.
- Gorbalenya AE, Baker SC, Baric RS, et al. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology 2020; 5(4): 536-44.
- World Health Organization. Rolling updates on coronavirus disease (COVID-19). Available at: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen. Accessed 30 April 2020.
- U.S. Food & Drug Administration. Coronavirus Disease 2019 (COVID-19) Emergency Use Authorizations for Medical Devices. Available at: https://www.fda.gov/medical-devices/emergency-use-authorizations-medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices. Accessed 7 March 2023.
- U.S. Food & Drug Administration. At-Home OTC COVID-19 Diagnostic Tests. Available at: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/home-otc-covid-19-diagnostic-tests#list. Accessed 3 March 2023.
- U.S. Food and Drug Administration. In Vitro Diagnostics EUAs - Molecular Diagnostic Tests for SARS-CoV-2. Available at: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2. Accessed 11 December 2022.
- Centers for Disease Control and Prevention. Symptoms of Coronavirus. Available at: https://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html. Accessed 3 May 2020.
- Burke RM, Killerby ME, Newton S, et al. Symptom Profiles of a Convenience Sample of Patients with COVID-19 — United States, January–April 2020. Morbidity and Mortality Weekly Report - CDC 2020; 69(28): 904-8.
- Morgan RL, Florez I, Falavigna M, et al. Development of rapid guidelines: 3. GIN-McMaster Guideline Development Checklist extension for rapid recommendations. Health Res Policy Syst 2018; 16(1): 63.
- Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 2. Framing the question and deciding on important outcomes. J Clin Epidemiol 2011; 64(4): 395-400.
- Whiting PF, Rutjes AW, Westwood ME, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med 2011; 155(8): 529-36.
- Schünemann HJ, Mustafa RA, Brozek J, et al. GRADE guidelines: 21 part 1. Study design, risk of bias, and indirectness in rating the certainty across a body of evidence for test accuracy. J Clin Epidemiol 2020; 122: 129-41.
- Schünemann HJ, Mustafa RA, Brozek J, et al. GRADE guidelines: 21 part 2. Test accuracy: inconsistency, imprecision, publication bias, and other domains for rating the certainty of evidence and presenting it in evidence profiles and summary of findings tables. J Clin Epidemiol 2020; 122: 142-52.
- Ai J-W, Zhang H-C, Xu T, et al. Optimizing diagnostic strategy for novel coronavirus pneumonia, a multi-center study in Eastern China. 2020: 2020.02.13.20022673.
- Alexander PE, Bero L, Montori VM, et al. World Health Organization recommendations are often strong based on low confidence in effect estimates. J Clin Epidemiol 2014; 67(6): 629-34.
- Guyatt GH, Alonso-Coello P, Schünemann HJ, et al. Guideline panels should seldom make good practice statements: guidance from the GRADE Working Group. J Clin Epidemiol 2016; 80: 3-7.
- Altamimi AMO, D. A.: Alaifan, T. A.: Taha, M. T.: Alhothali, M. T.: Alzahrani, F. A.: Albarrag, A. M. Assessment of 12 qualitative RT-PCR commercial kits for the detection of SARS-CoV-2. J Med Virol 2021; 93(5): 3219-26.
- Kim HN, Yoon SY, Lim CS, Yoon J. Comparison of three molecular diagnostic assays for SARS-CoV-2 detection: Evaluation of analytical sensitivity and clinical performance. Journal of Clinical Laboratory Analysis 2022.
- Lephart PRB, M. A.: LeBar, W.: McClellan, S.: Barron, K.: Schroeder, L.: Newton, D. W. Comparative study of four SARS-CoV-2 Nucleic Acid Amplification Test (NAAT) platforms demonstrates that ID NOW performance is impaired substantially by patient and specimen type. Diagn Microbiol Infect Dis 2021; 99(1): 115200.
- Yun J, Park JH, Kim N, et al. Evaluation of Three Multiplex Real-time Reverse Transcription PCR Assays for Simultaneous Detection of SARS-CoV-2, Influenza A/B, and Respiratory Syncytial Virus in Nasopharyngeal Swabs. J Korean Med Sci 2021; 36(48): e328.
- Smith E, Zhen W, Manji R, Schron D, Duong S, Berry GJ. Analytical and Clinical Comparison of Three Nucleic Acid Amplification Tests for SARS-CoV-2 Detection. J Clin Microbiol 2020; 58(9): e01134-20.
- Centers for Disease Control and Prevention. Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens for COVID-19. Available at: https://www.cdc.gov/coronavirus/2019-ncov/lab/guidelines-clinical-specimens.html. Accessed 22 April 2020.
- U.S. Food and Drug Administration. FAQs on Testing for SARS-CoV-2. Available at: https://www.fda.gov/medical-devices/emergency-situations-medical-devices/faqs-diagnostic-testing-sars-cov-2. Accessed 29 April 2020.
- Echavarria M, Reyes NS, Rodriguez PE, et al. Self-collected saliva for SARS-CoV-2 detection: A prospective study in the emergency room. J Med Virol 2021; 93(5): 3268-72.
- Vos MB, Gonzalez MD, Stone C, et al. Comparison of Mid-turbinate Nasal Swabs, Saliva, and Nasopharyngeal Swabs for SARS-CoV-2 Reverse Transcription-Polymerase Chain Reaction Testing in Pediatric Outpatients. Arch Pathol Lab Med 2022; 146(9): 1056-61.
- Rao M, Rashid FA, Sabri F, et al. Comparing Nasopharyngeal Swab and Early Morning Saliva for the Identification of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis 2021; 72(9): e352-e6.
- Landry ML, Criscuolo J, Peaper DR. Challenges in use of saliva for detection of SARS CoV-2 RNA in symptomatic outpatients. J Clin Virol 2020; 130: 104567.
- Procop GW, Shrestha NK, Vogel S, et al. A Direct Comparison of Enhanced Saliva to Nasopharyngeal Swab for the Detection of SARS-CoV-2 in Symptomatic Patients. J Clin Microbiol 2020; 58(11).
- Procop GW, Brock JE, Reineks EZ, et al. A Comparison of Five SARS-CoV-2 Molecular Assays With Clinical Correlations. Am J Clin Pathol 2020.
- McCormick-Baw C, Morgan K, Gaffney D, et al. Saliva as an Alternate Specimen Source for Detection of SARS-CoV-2 in Symptomatic Patients Using Cepheid Xpert Xpress SARS-CoV-2. J Clin Microbiol 2020; 58(8): e01109-20.
- Sun Q, Li J, Ren H, et al. Saliva as a testing specimen with or without pooling for SARS-CoV-2 detection by multiplex RT-PCR test. PLoS One 2021; 16(2): e0243183.
- Balaska S, Pilalas D, Takardaki A, et al. Evaluation of the Advanta Dx SARS-CoV-2 RT-PCR Assay, a High-Throughput Extraction-Free Diagnostic Test for the Detection of SARS-CoV-2 in Saliva: A Diagnostic Accuracy Study. Diagnostics (Basel) 2021; 11(10).
- Banerjee D, Sasidharan A, Abdulhamid A, et al. Diagnostic Yield of Saliva for SARS-CoV-2 Molecular Testing in Children. J Pediatric Infect Dis Soc 2021; 10(10): 967-9.
- Hanson KE, Barker AP, Hillyard DR, et al. Self-Collected Anterior Nasal and Saliva Specimens versus Healthcare Worker-Collected Nasopharyngeal Swabs for the Molecular Detection of SARS-CoV-2. J Clin Microbiol 2020; 58(11): e01824-20.
- Kandel C, Zheng J, McCready J, et al. Detection of SARS-CoV-2 from Saliva as Compared to Nasopharyngeal Swabs in Outpatients. Viruses 2020; 12(11).
- Nacher M, Mergeay-Fabre M, Blanchet D, et al. Diagnostic accuracy and acceptability of molecular diagnosis of COVID-19 on saliva samples relative to nasopharyngeal swabs in tropical hospital and extra-hospital contexts: The COVISAL study. PLoS One 2021; 16(9): e0257169.
- Iwasaki S, Fujisawa S, Nakakubo S, et al. Comparison of SARS-CoV-2 detection in nasopharyngeal swab and saliva. J Infect 2020; 81(2): e145-e7.
- Sogbesan K, Sogbesan T, Hata DJ, et al. Use of Self-Collected Saliva Samples for the Detection of SARS-CoV-2. Lab Med 2022; 53(6): 580-4.
- Fougere Y, Schwob JM, Miauton A, et al. Performance of RT-PCR on Saliva Specimens Compared With Nasopharyngeal Swabs for the Detection of SARS-CoV-2 in Children: A Prospective Comparative Clinical Trial. Pediatr Infect Dis J 2021; 40(8): e300-e4.
- Bhattacharya DP, D.: Rout, U. K.: Dash, P.: Nanda, R. R.: Dash, G. C.: Kanungo, S.: Palo, S. K.: Giri, S.: Choudhary, H. R.: Kshatri, J. S.: Turuk, J.: Mishra, B. K.: Lenka, R. K.: Dash, S.: Pati, S. Saliva for diagnosis of SARS-CoV-2: First report from India. J Med Virol 2021; 93(4): 2529-33.
- Uršič T, Kogoj R, Šikonja J, et al. Performance of nasopharyngeal swab and saliva in detecting Delta and Omicron SARS-CoV-2 variants. Journal of Medical Virology 2022.
- Al Suwaidi H, Senok A, Varghese R, et al. Saliva for molecular detection of SARS-CoV-2 in school-age children. Clinical Microbiology and Infection 2021; 27(9): 1330-5.
- Altawalah H, AlHuraish F, Alkandari WA, Ezzikouri S. Saliva specimens for detection of severe acute respiratory syndrome coronavirus 2 in Kuwait: A cross-sectional study. J Clin Virol 2020; 132: 104652.
- Masse S, Bonnet C, Vilcu AM, et al. Are Posterior Oropharyngeal Saliva Specimens an Acceptable Alternative to Nasopharyngeal Sampling for the Monitoring of SARS-CoV-2 in Primary-Care Settings? Viruses 2021; 13(5).
- LeGoff J, Kernéis S, Elie C, et al. Evaluation of a saliva molecular point of care for the detection of SARS-CoV-2 in ambulatory care. Sci Rep 2021; 11(1): 21126.
- Pitman JL, Morris AJ, Grice S, et al. Validation of a molecular assay to detect SARS-CoV-2 in saliva. New Zealand Medical Journal 2021; 134(1547): 14-27.
- Migueres M, Mengelle C, Dimeglio C, et al. Saliva sampling for diagnosing SARS-CoV-2 infections in symptomatic patients and asymptomatic carriers. J Clin Virol 2020; 130: 104580.
- Migueres M, Vellas C, Abravanel F, et al. Testing individual and pooled saliva samples for sars-cov-2 nucleic acid: a prospective study. Diagn Microbiol Infect Dis 2021; 101(3): 115478.
- Patel MR, Carroll D, Ussery E, et al. Performance of Oropharyngeal Swab Testing Compared With Nasopharyngeal Swab Testing for Diagnosis of Coronavirus Disease 2019-United States, January 2020-February 2020. Clin Infect Dis 2021; 72(3): 403-10.
- Pham J, Meyer S, Nguyen C, et al. Performance Characteristics of a High-Throughput Automated Transcription-Mediated Amplification Test for SARS-CoV-2 Detection. J Clin Microbiol 2020; 58(10): e01669-20.
- Wang X, Tan L, Wang X, et al. Comparison of nasopharyngeal and oropharyngeal swabs for SARS-CoV-2 detection in 353 patients received tests with both specimens simultaneously. Int J Infect Dis 2020; 94: 107-9.
- Kandel CE, Young M, Serbanescu MA, et al. Detection of severe acute respiratory coronavirus virus 2 (SARS-CoV-2) in outpatients: A multicenter comparison of self-collected saline gargle, oral swab, and combined oral-anterior nasal swab to a provider collected nasopharyngeal swab. Infection Control and Hospital Epidemiology 2021; 42(11): 1340-4.
- Harrington A, Cox B, Snowdon J, et al. Comparison of Abbott ID Now and Abbott m2000 Methods for the Detection of SARS-CoV-2 from Nasopharyngeal and Nasal Swabs from Symptomatic Patients. J Clin Microbiol 2020; 58(8): e00798-20.
- Callahan C, Lee RA, Lee GR, Zulauf K, Kirby JE, Arnaout R. Nasal swab performance by collection timing, procedure, and method of transport for patients with sars-cov-2. Journal of Clinical Microbiology 2021; 59(9).
- Montano MA, Bemer MJ, Heller KB, et al. Performance of anterior nares and tongue swabs for nucleic acid, Nucleocapsid, and Spike antigen testing for detecting SARS-CoV-2 against nasopharyngeal PCR and viral culture. Int J Infect Dis 2022; 117: 287-94.
- Tu YP, Jennings R, Hart B, et al. Swabs Collected by Patients or Health Care Workers for SARS-CoV-2 Testing. N Engl J Med 2020; 383(5): 494-6.
- LeBlanc JJ, Heinstein C, MacDonald J, Pettipas J, Hatchette TF, Patriquin G. A combined oropharyngeal/nares swab is a suitable alternative to nasopharyngeal swabs for the detection of SARS-CoV-2. J Clin Virol 2020; 128: 104442.
- Sahni LC, Avadhanula V, Ortiz CS, et al. Comparison of Mid-Turbinate and Nasopharyngeal Specimens for Molecular Detection of SARS-CoV-2 among Symptomatic Outpatients at a Pediatric Drive-Through Testing Site. Journal of the Pediatric Infectious Diseases Society 2021; 10(8): 872-9 %9 Article %! Comparison of Mid-Turbinate and Nasopharyngeal Specimens for Molecular Detection of SARS-CoV-2 among Symptomatic Outpatients at a Pediatric Drive-Through Testing Site %@ 2048-7207 2048-7193.
- Vermeiren C, Marchand-Senécal X, Sheldrake E, et al. Comparison of Copan ESwab and FLOQSwab for COVID-19 Diagnosis: Working around a Supply Shortage. J Clin Microbiol 2020; 58(6): e00669-20.
- McCulloch DJ, Kim AE, Wilcox NC, et al. Comparison of Unsupervised Home Self-collected Midnasal Swabs With Clinician-Collected Nasopharyngeal Swabs for Detection of SARS-CoV-2 Infection. JAMA Netw Open 2020; 3(7): e2016382.
- Tu Y-P, Jennings R, Hart B, et al. Patient-collected tongue, nasal, and mid-turbinate swabs for SARS-CoV-2 yield equivalent sensitivity to health care worker collected nasopharyngeal swabs. 2020: 2020.04.01.20050005.
- Péré H, Podglajen I, Wack M, et al. Nasal Swab Sampling for SARS-CoV-2: a Convenient Alternative in Times of Nasopharyngeal Swab Shortage. Journal of Clinical Microbiology 2020; 58(6): e00721-20.
- Kocagoz T, Can O, Yurttutan Uyar N, et al. Simple concentration method enables the use of gargle and mouthwash instead of nasopharyngeal swab sampling for the diagnosis of COVID-19 by PCR. European Journal of Clinical Microbiology and Infectious Diseases 2021; 40(12): 2617-22.
- Babady NE, McMillen T, Jani K, et al. Performance of Severe Acute Respiratory Syndrome Coronavirus 2 Real-Time RT-PCR Tests on Oral Rinses and Saliva Samples. J Mol Diagn 2021; 23(1): 3-9.
- Zander J, Scholtes S, Ottinger M, et al. Self-Collected Gargle Lavage Allows Reliable Detection of SARS-CoV-2 in an Outpatient Setting. Microbiology Spectrum 2021; 9(1): 1-5.
- Laferl H, Seitz T, Baier-Grabner S, et al. Evaluation of RT-qPCR of mouthwash and buccal swabs for detection of SARS-CoV-2 in children and adults. American Journal of Infection Control 2021.
- Rattan A, Joerger J, Williams D, Pollock NR. Similar SARS-CoV-2 Ct value distributions in anterior nares versus nasopharyngeal samples from symptomatic children during Delta and Omicron surges. Journal of the Pediatric Infectious Diseases Society 2023; 12(2): 109-12.
- Kojima N, Turner F, Slepnev V, et al. Self-Collected Oral Fluid and Nasal Swabs Demonstrate Comparable Sensitivity to Clinician Collected Nasopharyngeal Swabs for Coronavirus Disease 2019 Detection. Clin Infect Dis 2021; 73(9): e3106-e9.
- Wehrhahn MC, Robson J, Brown S, et al. Self-collection: An appropriate alternative during the SARS-CoV-2 pandemic. J Clin Virol 2020; 128: 104417.
- Thwe PM, Ren P. How many are we missing with ID NOW COVID-19 assay using direct nasopharyngeal swabs? Findings from a mid-sized academic hospital clinical microbiology laboratory. Diagn Microbiol Infect Dis 2020; 98(2): 115123.
- McDonald S, Courtney DM, Clark AE, et al. Diagnostic Performance of a Rapid Point-of-care Test for SARS-CoV-2 in an Urban Emergency Department Setting. Acad Emerg Med 2020; 27(8): 764-6.
- Barker KR, Small LN, Thai DV, Sohn KY, Rosella LC. Evaluating the Ability to ID (COVID-19) NOW: a Large Real-World Prospective Evaluation of the Abbott ID NOW COVID-19 Assay. Microbiol Spectr 2022; 10(3): e0051322.
- Farfour E, Asso-Bonnet M, Vasse M, et al. The ID NOW COVID-19, a high-speed high-performance assay. European Journal of Clinical Microbiology and Infectious Diseases 2021; 40(9): 2041-5.
- Cradic KL, M.: Ozbolt, P.: Fatica, L.: Landon, L.: Lieber, M.: Yang, D.: Swickard, J.: Wongchaowart, N.: Fuhrman, S.: Antonara, S. Clinical evaluation and utilization of multiple molecular in vitro diagnostic assays for the detection of SARS-CoV-2. American Journal of Clinical Pathology 2020; 154(2): 201-7.
- Hofman P, Boutros J, Benchetrit D, et al. A rapid near-patient RT-PCR test for suspected COVID-19: a study of the diagnostic accuracy. Ann Transl Med 2021; 9(11): 921.
- De Pace V, Caligiuri P, Ricucci V, et al. Rapid diagnosis of SARS-CoV-2 pneumonia on lower respiratory tract specimens. BMC Infect Dis 2021; 21(1): 926.
- Loeffelholz MJ, Alland D, Butler-Wu SM, et al. Multicenter Evaluation of the Cepheid Xpert Xpress SARS-CoV-2 Test. J Clin Microbiol 2020; 58(8): e00926-20.
- Moran A, Beavis KG, Matushek SM, et al. Detection of SARS-CoV-2 by Use of the Cepheid Xpert Xpress SARS-CoV-2 and Roche cobas SARS-CoV-2 Assays. J Clin Microbiol 2020; 58(8): e00772-20.
- Wolters F, van de Bovenkamp J, van den Bosch B, et al. Multi-center evaluation of cepheid xpert® xpress SARS-CoV-2 point-of-care test during the SARS-CoV-2 pandemic. J Clin Virol 2020; 128: 104426.
- Fitoussi F, Tonen-Wolyec S, Awaida N, Dupont R, Bélec L. Analytical performance of the point-of-care BIOSYNEX COVID-19 Ag BSS for the detection of SARS-CoV-2 nucleocapsid protein in nasopharyngeal swabs: a prospective field evaluation during the COVID-19 third wave in France. Infection 2021: 1-9.
- Mack CD, Wasserman EB, Hostler CJ, et al. Effectiveness and use of reverse transcriptase polymerase chain reaction point of care testing in a large-scale COVID-19 surveillance system. Pharmacoepidemiology and Drug Safety 2022.
- Visseaux B, Le Hingrat Q, Collin G, et al. Evaluation of the QIAstat-Dx Respiratory SARS-CoV-2 Panel, the First Rapid Multiplex PCR Commercial Assay for SARS-CoV-2 Detection. J Clin Microbiol 2020; 58(8): e00630-20.
- Renzoni AP, F.: Ngo Nsoga, M. T.: Yerly, S.: Boehm, E.: Gayet-Ageron, A.: Kaiser, L.: Schibler, M. Analytical evaluation of visby medical rt-pcr portable device for rapid detection of sars-cov-2. Diagnostics 2021; 11(5).
- Challener DW, Shah A, O'Horo JC, Berbari E, Binnicker MJ, Tande AJ. Low Utility of Repeat Real-Time PCR Testing for SARS-CoV-2 in Clinical Specimens. Mayo Clin Proc 2020; 95(9): 1942-5.
- Long DR, Gombar S, Hogan CA, et al. Occurrence and Timing of Subsequent Severe Acute Respiratory Syndrome Coronavirus 2 Reverse-transcription Polymerase Chain Reaction Positivity Among Initially Negative Patients. Clin Infect Dis 2021; 72(2): 323-6.
- Ramachandran A, Noble J, Deucher A, Miller S, Tang PW, Wang RC. Performance of Abbott ID-Now rapid nucleic amplification test for laboratory identification of COVID-19 in asymptomatic emergency department patients. J Am Coll Emerg Physicians Open 2021; 2(6): e12592.
- Micocci M, Gordon AL, Seo MK, et al. Is point-of-care testing feasible and safe in care homes in England? An exploratory usability and accuracy evaluation of a point-of-care polymerase chain reaction test for SARS-CoV-2. Age and Ageing 2021; 50(5): 1464-72 %9 Article %! Is point-of-care testing feasible and safe in care homes in England? An exploratory usability and accuracy evaluation of a point-of-care polymerase chain reaction test for SARS-CoV-2 %@ 8-2834 0002-729.
- Antonara S, Ozbolt P, Landon L, et al. Detection of SARS-CoV-2 infection in asymptomatic populations using the DiaSorin molecular Simplexa and Roche Cobas EUA assays. Diagn Microbiol Infect Dis 2022; 102(1): 115513.
- Freire-Paspuel B, Garcia-Bereguiain MA. Analytical and Clinical Evaluation of "AccuPower SARS-CoV-2 Multiplex RT-PCR kit (Bioneer, South Korea)" and "Allplex 2019-nCoV Assay (Seegene, South Korea)" for SARS-CoV-2 RT-PCR Diagnosis: Korean CDC EUA as a Quality Control Proxy for Developing Countries. Front Cell Infect Microbiol 2021; 11: 630552.
- Freire-Paspuel B, Vega-Marino P, Velez A, Cruz M, Perez F, Garcia-Bereguiain MA. Analytical and clinical comparison of Viasure (CerTest Biotec) and 2019-nCoV CDC (IDT) RT-qPCR kits for SARS-CoV2 diagnosis. Virology 2021; 553: 154-6.
- Hann A, Flemming S, Reimer S, et al. Impact of pre-procedural testing on SARS-CoV-2 transmission to endoscopy staff. Gut 2022; 71(11): 2167-9.
- Podboy A, Cholankeril G, Cianfichi L, Guzman E, Jr., Ahmed A, Banerjee S. Implementation and Impact of Universal Preprocedure Testing of Patients for COVID-19 Before Endoscopy. Gastroenterology 2020; 159(4): 1586-8 e4.
- Hayee B, Hussain, East J, Rees CJ, Penman I. Multicentre prospective study of COVID-19 transmission following outpatient GI endoscopy in the UK. Gut 2021; 70(5): 825.
- Hayee B, Hussain, Bhandari P, Rees CJ, Penman I. COVID-19 transmission following outpatient endoscopy during pandemic acceleration phase involving SARS-CoV-2 VOC 202012/01 variant in UK. Gut 2021; 70(12): 2227.
- Repici A, Aragona G, Cengia G, et al. Low risk of COVID-19 transmission in GI endoscopy. Gut 2020; 69(11): 1925.
- Jagannath S, Agarwal A, Gunjan D, et al. Mandatory preprocedure testing for SARS-CoV-2 for all-comers may not be required for resuming endoscopic services amidst the ongoing COVID-19 pandemic. Gut 2021; 70(9): 1805.
- Khorrami Minaei S, Garrido Durán C, García Hernández M, García Amengual I, Mena Ribas A. Poor agreement between clinical screening and universal pre-procedure SARS-CoV-2 PCR testing prior to endoscopy. Rev Esp Enferm Dig 2021; 113(9): 649-55.
- American Society of Transplantation. COVID-19: FAQs for Organ Transplantation Available at: https://www.myast.org/sites/default/files/COVID%20FAQ%20for%20Tx%20professionals%202-2023%20FINAL.pdf. Accessed 1 July 2023.
- Jonker PKC, van der Plas WY, Steinkamp PJ, et al. Perioperative SARS-CoV-2 infections increase mortality, pulmonary complications, and thromboembolic events: A Dutch, multicenter, matched-cohort clinical study. Surgery 2021; 169(2): 264-74.
- Collaborative CO. Outcomes and Their State-level Variation in Patients Undergoing Surgery With Perioperative SARS-CoV-2 Infection in the USA: A Prospective Multicenter Study. Ann Surg 2022; 275(2): 247-51.
- Collaborative C, Collaborative G. Timing of surgery following SARS-CoV-2 infection: an international prospective cohort study. Anaesthesia 2021; 76(6): 748-58.
- Le ST, Kipnis P, Cohn B, Liu VX. COVID-19 Vaccination and the Timing of Surgery Following COVID-19 Infection. Ann Surg 2022; 276(5): e265-e72.
- Deng JZ, Chan JS, Potter AL, et al. The Risk of Postoperative Complications After Major Elective Surgery in Active or Resolved COVID-19 in the United States. Ann Surg 2022; 275(2): 242-6.
- Bryant JM, Boncyk CS, Rengel KF, et al. Association of Time to Surgery After COVID-19 Infection With Risk of Postoperative Cardiovascular Morbidity. JAMA Netw Open 2022; 5(12): e2246922.
- COVIDSurg Collaborative. Mortality and pulmonary complications in patients undergoing surgery with perioperative SARS-CoV-2 infection: an international cohort study. Lancet 2020; 396(10243): 27-38.
- Doglietto F, Vezzoli M, Gheza F, et al. Factors Associated With Surgical Mortality and Complications Among Patients With and Without Coronavirus Disease 2019 (COVID-19) in Italy. JAMA Surg 2020; 155(8): 691-702.
- Mack CD, Wasserman EB, Killerby ME, et al. Results from a Test-to-Release from Isolation Strategy Among Fully Vaccinated National Football League Players and Staff Members with COVID-19 - United States, December 14-19, 2021. MMWR Morb Mortal Wkly Rep 2022; 71(8): 299-305.
- Chu VT, Schwartz NG, Donnelly MAP, et al. Comparison of Home Antigen Testing With RT-PCR and Viral Culture During the Course of SARS-CoV-2 Infection. JAMA Internal Medicine 2022; 182(7): 701-9.
- Aranha C, Patel V, Bhor V, Gogoi D. Cycle threshold values in RT-PCR to determine dynamics of SARS-CoV-2 viral load: An approach to reduce the isolation period for COVID-19 patients. J Med Virol 2021; 93(12): 6794-7.
- Hakki S, Zhou J, Jonnerby J, et al. Onset and window of SARS-CoV-2 infectiousness and temporal correlation with symptom onset: a prospective, longitudinal, community cohort study. Lancet Respir Med 2022; 10(11): 1061-73.
- U.S. Food & Drug Administration. FDA Permits Marketing of First SARS-CoV-2 Diagnostic Test Using Traditional Premarket Review Process. Available at: https://www.fda.gov/news-events/press-announcements/fda-permits-marketing-first-sars-cov-2-diagnostic-test-using-traditional-premarket-review-process. Accessed 5 March 2023.
- U.S. Department of Health and Human Services. Fact Sheet: COVID-19 Public Health Emergency Transition Roadmap. Available at: https://www.hhs.gov/about/news/2023/02/09/fact-sheet-covid-19-public-health-emergency-transition-roadmap.html#:~:text=Based%20on%20current%20COVID%2D19,day%20on%20May%2011%2C%202023. Accessed 3 March 2023.
- U.S. Food & Drug Administration. FAQs: What happens to EUAs when a public health emergency ends? Available at: https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/faqs-what-happens-euas-when-public-health-emergency-ends. Accessed 15 March 2023.
Supplementary Information
- Figure s1. PRISMA Flow Diagram
- Table s1. PICO Questions Identified by the Panel Through the Three Guideline Versions
- Table s2. Search Strategy
- Table s3. QUADAS-2 Risk of Bias Assessment
- Table s4. Baseline Characteristics of the Included Studies from Systematic Review
- Table s5. Narrative Summaries of Included Studies from Additional Targeted Searches
Supplement B: Recommendation 1
- Figure s2a. Forest Plot for the Sensitivity for Molecular Tests
- Figure s2b. Forest Plot for the Specificity for Molecular Tests
Supplement C: Recommendation 2
- Table s6. GRADE Evidence Profile of Molecular Test Accuracy Results Based on Different Sample Sites Using a Prevalence/Pre-Test Probability of 5%
- Table s7. GRADE Evidence Profile of Molecular Test Accuracy Results Based on Different Sample Sites Using a Prevalence/Pre-Test Probability of 50%
- Figure s3a. Forest Plot for the Sensitivity for Saliva (Overall) vs Nasopharyngeal (NP)
- Figure s3b. Forest Plot for the Specificity for Saliva (Overall) vs Nasopharyngeal (NP)
- Figure s4a. Forest Plot for the Sensitivity for Saliva (Pediatrics) vs Nasopharyngeal (NP)
- Figure s4b. Forest Plot for the Specificity for Saliva (Pediatrics) vs Nasopharyngeal (NP)
- Figure s5a. Forest Plot for the Sensitivity for Saliva (Without Cough) vs Nasopharyngeal (NP)
- Figure s5b. Forest Plot for the Specificity for Saliva (Without Cough) vs Nasopharyngeal (NP)
- Figure s6a. Forest Plot for the Sensitivity for Saliva (With Cough) vs Nasopharyngeal (NP)
- Figure s6b. Forest Plot for the Specificity for Saliva (With Cough) vs Nasopharyngeal (NP)
- Figure s7a. Forest Plot for the Sensitivity for Oropharyngeal (OP) vs Nasopharyngeal (NP)
- Figure s7b. Forest Plot for the Specificity for Oropharyngeal (OP) vs Nasopharyngeal (NP)
- Figure s8a. Forest Plot for the Sensitivity for Anterior Nasal (AN) vs Nasopharyngeal (NP)
- Figure s8b. Forest Plot for the Specificity for Anterior Nasal (AN) vs Nasopharyngeal (NP)
- Figure s9a. Forest Plot for the Sensitivity for Combined Oropharyngeal (OP) and Anterior Nasal (AN) vs Nasopharyngeal (NP)
- Figure s9b. Forest Plot for the Specificity for Combined Oropharyngeal (OP) and Anterior Nasal (AN) vs Nasopharyngeal (NP)
- Figure s10a. Forest Plot for the Sensitivity for Mid-Turbinate (Mt) vs Nasopharyngeal (NP)
- Figure s10b. Forest Plot for the Specificity for Mid-Turbinate (MT) vs Nasopharyngeal (NP)
- Figure s11a. Forest Plot for the Sensitivity for Mouth Gargle vs Nasopharyngeal (NP)
- Figure s11b. Forest Plot for the Specificity for Mouth Gargle vs Nasopharyngeal (NP)
Supplement D: Recommendation 3
- Figure s12a. Forest Plot for the Sensitivity of Self-Collected Nasal Sampling with Reference Being Any Positive Test (HCW Or Self Collected)
- Figure s12b. Forest Plot for the Specificity of Self-Collected Nasal Sampling with Reference Being Any Positive Test (HCW Or Self Collected)
- Figure s13a. Forest Plot for the Sensitivity of Health Care Worker (HCW)-Collected Nasal Sampling with Reference Being Any Positive Test (HCW Or Self Collected)
- Figure s13b. Forest Plot for the Specificity Of Health Care Worker (HCW)-Collected Nasal Sampling with Reference Being Any Positive Test (HCW Or Self Collected)
Supplement E: Recommendation 5
- Figure s14a. Sensitivity of Rapid vs Laboratory-Based NAAT Accuracy in All Studies (Overall)
- Figure s14b. Specificity of Rapid vs Standard Laboratory-Based NAAT Accuracy in All Studies (Overall)
- Figure s15a. Sensitivity of Rapid vs Standard Laboratory-Based NAAT Accuracy in Symptomatic Individuals Only
- Figure s15b. Specificity of Rapid vs Standard Laboratory-Based NAAT Accuracy in Symptomatic Individuals Only
Supplement F: Recommendation 8
- Figure s16a. Sensitivity of Rapid vs Standard Laboratory-Based Naats Accuracy in Asymptomatic Individuals Only
- Figure s16b. Specificity of Rapid vs Standard Laboratory-Based Naats Accuracy in Asymptomatic Individuals Only
Additional Resources
Food and Drug Administration: Specimen Collection FAQs
New England Journal of Medicine: How to Obtain a Nasopharyngeal Swab Specimen