
IDSA Guidelines on the Treatment and Management of Patients with COVID-19
Published by IDSA, 5/27/2021. Last updated,
COVID-19 Guideline, Part 2: Infection Prevention
COVID-19 Guideline, Part 3: Molecular Testing
COVID-19 Guideline, Part 4: Serologic Testing
COVID-19 Guideline, Part 5: Antigen Testing
Management of Drug Interactions With Nirmatrelvir/Ritonavir (Paxlovid®): Resource for Clinicians
Adarsh Bhimraj,* Rebecca L. Morgan,** Amy Hirsch Shumaker, Lindsey Baden, Vincent Chi-Chung Cheng, Kathryn M. Edwards, Jason C. Gallagher, Rajesh T. Gandhi, William J. Muller, Mari M. Nakamura, John C. O’Horo, Robert W. Shafer, Shmuel Shoham, M. Hassan Murad,** Reem A. Mustafa,** Shahnaz Sultan,** Yngve Falck-Ytter**
*Corresponding Author **Methodologist
June 26, 2023
Version 11.0.0 has been released and includes the following:
- Convalescent Plasma: A new recommendation was developed against the routine use of convalescent plasma among immunocompromised patients hospitalized with COVID-19. Additionally, this section includes updated remarks for the existing recommendation on the use of convalescent plasma for ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options.
- Anakinra: This section has been added and includes a new recommendation against the routine use of anakinra among hospitalized patients with severe COVID-19.
- Nirmatrelvir/Ritonavir: This section includes updated remarks for the existing recommendation on the use of nirmatrelvir/ritonavir for ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease.
This update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
Update History
April 11, 2023
The following recommendation was updated based on newly available literature and approvals. It was provided here for immediate use and was later integrated into the website on June 26, 2023 as part of Version 11.0.0.
- Convalescent Plasma (PDF): A new recommendation was developed against the routine use of convalescent plasma among immunocompromised patients hospitalized with COVID-19. Additionally, this section includes updated remarks for the existing recommendation on the use of convalescent plasma for ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options.
This update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
March 14, 2023
Version 10.2.1 has been released and includes updated evidence summaries and clarified remarks on the use of molnupiravir. No changes have been made to the current recommendation.
January 20, 2023
The following recommendations were updated based on newly available literature and approvals. They were provided here for immediate use and were later integrated into the website on February 8, 2023 as part of Version 10.2.0.
- Neutralizing Antibodies for Pre-Exposure Prophylaxis: A remark was added to the recommendation regarding resistance of tixagevimab/cilgavimab (Evusheld) in the US. The agent has an Emergency Use Authorization by the US FDA and may be used in other parts of the world where the circulating COVID-19 variants may still be susceptible to it.
- Neutralizing Antibodies for Post-Exposure Prophylaxis: This recommendation was retired and replaced with a statement mentioning in vitro resistance of casirivimab/imdevimab to circulating strains of COVID-19 in the US.
- Neutralizing Antibodies for Treatment: This recommendation was retired and replaced with a statement mentioning that the US FDA withdrew Emergency Use Authorization for bebtelovimab, the one anti-SARS CoV-2 neutralizing antibody product that had retained in vitro activity against most previously circulating SARS-CoV-2 variants, leaving no available neutralizing antibody product in the United Sates for treatment of COVID-19.
This update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
November 21, 2022
Version 10.1.1 has been released and includes updates to Figure 7, Figure 8, and Figure 9.
November 15, 2022
Version 10.1.0 has been released and includes the following:
- Inhaled Corticosteroids: This recommendation on the use of inhaled corticosteroids among ambulatory patients with mild-to-moderate COVID-19 has been revised.
- Ivermectin: This section has been updated based on newly added literature.
This update been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
November 2, 2022
The following sections have been updated based on newly available literature and approvals. They were provided here for immediate use and were integrated into the website as part of Version 10.1.0
- Ivermectin: This section has been updated based on newly added literature.
- Inhaled Corticosteroids: This recommendation on the use of inhaled corticosteroids among ambulatory patients with mild-to-moderate COVID-19 has been revised.
This update been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
October 18, 2022
Version 10.0.1 has been released and includes minor corrections to Table 22 and Table 35.
August 30, 2022
The following sections were added/revised based on newly available literature and/or approvals. They were provided here for immediate use and have now been integrated into the website as part of Version 10.0.0.
- Ivermectin: Revised recommendations on the use of ivermectin in hospitalized and ambulatory persons with COVID-19.
- Colchicine: New recommendations on the use of colchicine in hospitalized and ambulatory persons with COVID-19.
Additionally, three new narrative sections have been developed:
- How to Approach a Patient when Considering Pharmacologic Treatments for COVID-19
- Bacterial Co-Infections and Antibiotic Use
- Pediatric Considerations for Treatment of SARS-CoV-2 Infection and Multisystem Inflammatory Syndrome in Children
This update been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
July 12, 2022
Recommendations on the use of ivermectin in hospitalized and ambulatory persons with COVID-19 were revised. These revised recommendations were provided for immediate use and were later integrated into the website on August 30, 2022, as part of Version 10.0.0. This update has been endorsed by the Pediatric Infectious Diseases Society and the Society for Healthcare Epidemiology of America.
July 7, 2022
Two new recommendations on the use of colchicine in hospitalized and ambulatory persons with COVID-19 were developed. These new recommendations were provided for immediate use and were later integrated into the website on August 30, 2022, as part of Version 10.0.0. This update has been endorsed by the Pediatric Infectious Diseases Society and the Society for Healthcare Epidemiology of America.
June 29, 2022
Version 9.0.1 has been released and includes a footnote regarding ambulatory patients receiving convalescent plasma who have no other treatment options.
June 10, 2022
Version 9.0.0 has been released and includes the following:
- Famotidine: New recommendation on the use of famotidine in ambulatory patients with mild-to-moderate COVID-19; revised recommendation on the use of famotidine in hospitalized patients with severe COVID-19.
- Neutralizing Antibodies for Pre- and Post-Exposure Prophylaxis: Revised recommendation on the use of tixagevimab/cilgavimab as pre-exposure prophylaxis in moderately or severely immunocompromised individuals at increased risk for inadequate immune response to COVID-19 vaccine, OR for persons for whom the COVID-19 vaccine is not recommended due to a documented serious adverse reaction to the vaccine; revised recommendation on the use of casirivimab/imdevimab as post-exposure prophylaxis for persons exposed to COVID-19 at high risk of progression to severe disease.
- Neutralizing Antibodies for Treatment: Revised recommendation on the use of monoclonal antibodies in ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease.
- Janus Kinase Inhibitors (Baricitinib): Revised recommendation on the use of baricitinib with corticosteroids for hospitalized adults with severe COVID-19.
This update has been endorsed by the Society for Healthcare Epidemiology of America.
May 27, 2022
The following recommendation sections were added/revised based on newly available literature and/or approvals. They were provided here for immediate use and were later integrated into the website on June 10, 2022 as part of Version 9.0.0. These updates have been endorsed by the Society for Healthcare Epidemiology of America.
- Famotidine: New recommendation on the use of famotidine in ambulatory patients with mild-to-moderate COVID-19; revised recommendation on the use of famotidine in hospitalized patients with severe COVID-19.
- Neutralizing Antibodies for Pre- and Post-Exposure Prophylaxis: Revised recommendation on the use of tixagevimab/cilgavimab as pre-exposure prophylaxis in moderately or severely immunocompromised individuals at increased risk for inadequate immune response to COVID-19 vaccine, OR for persons for whom the COVID-19 vaccine is not recommended due to a documented serious adverse reaction to the vaccine; revised recommendation on the use of casirivimab/imdevimab as post-exposure prophylaxis for persons exposed to COVID-19 at high risk of progression to severe disease.
- Neutralizing Antibodies for Treatment: Revised recommendation on the use of monoclonal antibodies in ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease.
May 10, 2022
A recommendation on the use of baricitinib with corticosteroids for hospitalized adults with severe COVID-19 was revised. This revised recommendation was provided for immediate use and was later integrated into the website on June 10, 2022 as part of Version 9.0.0. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society.
March 23, 2022
Version 8.0.0 has been released and includes new recommendations on the use of inhaled glucocorticoids in ambulatory patients with mild-to-moderate COVID-19 and bebtelovimab in ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society.
March 18, 2022
A new recommendation was developed on the use of inhaled corticosteroids in ambulatory patients with mild-to-moderate COVID-19. This new recommendation was originally provided for immediate use and was later integrated into the website on March 23, 2022 as part of Version 8.0.0.
March 14, 2022
Version 7.0.1 has been released and includes an update to the dosing for tixagevimab/cilgavimab as pre-exposure prophylaxis for moderately or severely immunocompromised individuals at increased risk for inadequate immune response to COVID-19 vaccine OR for whom COVID-19 vaccine is not recommended due to a documented serious adverse reaction to the vaccine.
March 11, 2022
A new recommendation was developed on the use of bebtelovimab in ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease. This new recommendation was originally provided for immediate use and was later integrated into the website on March 23, 2022 as part of Version 8.0.0.
March 9, 2022
Version 7.0.0 has been released and includes new recommendations on the use of lopinavir/ritonavir for individuals exposed to or with COVID-19, a revised recommendation on the use of convalescent plasma in ambulatory patients with mild-to-moderate COVID-19, and a revised recommendation for the use of remdesivir in patients (ambulatory or hospitalized) with mild-to-moderate COVID-19 at high risk of progression to severe disease. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society.
February 22, 2022
Two new recommendations were developed on the use of lopinavir/ritonavir (prophylaxis for persons exposed to SARS-CoV-2; treatment for ambulatory patients with mild-to-moderate COVID-19). This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society. These new recommendations were originally provided for immediate use and were later integrated into the website on March 9, 2022 as part of Version 7.0.0.
February 16, 2022
A revised recommendation was released on the use of remdesivir in patients (ambulatory or hospitalized) with mild-to-moderate COVID at high risk for progression to severe disease. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society. This revised recommendation was originally provided for immediate use and was later integrated into the website on March 9, 2022 as part of Version 7.0.0.
February 8, 2022
A new recommendation was released on the use of convalescent plasma in ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease with no other treatment options. This recommendation was originally provided for immediate use and was later integrated into the website on March 9, 2022 as part of Version 7.0.0.
February 3, 2022
Version 6.0.2 has been released and includes an update to the evidence profile for nirmatrelvir/ritonavir in ambulatory patients (outcome of COVID-19 related hospitalizations).
January 18, 2022
Version 6.0.1 as been released and includes endorsement from the Society for Infectious Diseases Pharmacists.
January 12, 2022
Version 6.0.0 as been released and includes new recommendations on the use of remdesivir for ambulatory patients, tixagevimab/cilgavimab for pre-exposure prophylaxis, nirmatrelvir/ritonavir in ambulatory patients, and molnupiravir for ambulatory patients. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society.
December 30, 2021
A new recommendation was released on the use of molnupiravir for ambulatory patients with mild to moderate COVID-19 at high risk for progression to severe disease who have no other treatment options. This recommendation was originally provided for immediate use and was later integrated into the website on January 12, 2022 as part of Version 6.0.0.
December 24, 2021
The following recommendation sections were added based on newly available literature and/or approvals. They were provided here for immediate use and were later integrated into the website on January 12, 2022 as part of Version 6.0.0.
- Remdesivir: New recommendation on the use of remdesivir for ambulatory patients
- Neutralizing Antibodies for Prophylaxis: New recommendation on the use of tixagevimab/cilgavimab for pre-exposure prophylaxis in adults at increased risk for inadequate immune response to COVID-19 vaccine or for whom COVID-19 vaccine is not recommended
- Oral Antivirals: New recommendation on the use of nirmatrelvir/ritonavir in ambulatory patients with mild to moderate COVID-19 at high risk for progression to severe disease
- The language in the above section has been updated, with "nirmatrelvir/ritonavir" replacing "oral antivirals". Additionally, information on the use of brand name as well as use in patients hospitalized for reasons other than COVID-19 has been added.
November 18, 2021
Version 5.6.0 as been released and includes revised recommendations on the use of convalescent plasma in hospitalized and ambulatory patients with COVID-19; this update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
November 9, 2021
Version 5.5.3 has been released and contains a correction to the analysis for the outcome of mortality in ambulatory patients treated with fluvoxamine.
November 5, 2021
Version 5.5.2 has been released and includes updated literature for the use of fluvoxamine in ambulatory patients.
November 1, 2021
Version 5.5.1 has been released and includes endorsement from the Society of Infectious Diseases Pharmacists.
October 27, 2021
Version 5.5.0 has been released and contains a new recommendation on the use of fluvoxamine in ambulatory patients. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Pediatric Infectious Diseases Society.
October 18, 2021
Version 5.4.1 has been released and contains a minor correction to the neutralizing antibodies section. Subcutaneous has been removed to the dosing for bamlanivimab/etesevimab.
October 15, 2021
Version 5.4.0 has been released and includes revised remarks and a new evidence profile for the use of baricitinib for critically ill patients requiring invasive mechanical ventilation. This update has been endorsed by the Society of Infectious Diseases Pharmacists.
October 1, 2021
Version 5.3.1 has been released and contains a correction to the certainty of evidence for the new recommendation on the use of hydroxychloroquine as post-exposure prophylaxis. This recommendation has a moderate--not low--certainty of evidence.
September 30, 2021
Version 5.3.0 has been released and includes a new recommendation on the use of hydroxychloroquine as post-exposure prophylaxis. This update has been endorsed by the Society for Healthcare Epidemiology of America and the Society of Infectious Diseases Pharmacists.
September 24, 2021
Version 5.2.1 has been released and features existing recommendations on the use of neutralizing SARS-CoV-2 antibodies separated by prophylaxis vs. treatment.
September 21, 2021
Version 5.2.0 has been released and includes a new recommendation on the use of sarilumab. This update has been endorsed by the Society for Healthcare Epidemiology of America, the Pediatric Infectious Diseases Society, and the Society of Infectious Diseases Pharmacists.
September 14, 2021
Version 5.1.2 has been released and includes endorsement from the Society of Infectious Diseases Pharmacists.
September 3, 2021
Version 5.1.1 has been released and includes endorsement from the Pediatric Infectious Diseases Society.
August 27, 2021
Version 5.1.0 has been released and includes revised remarks for the use of baricitinib and new recommendations on the use of tofacitinib.
August 25, 2021
Version 5.0.0 has been released and includes new and updated recommendations for neutralizing antibodies and ivermectin. This update has been endorsed by the Society for Healthcare Epidemiology of America.
June 25, 2021
Version 4.4.1 has been released and includes endorsement from the Pediatric Infectious Diseases Society.
June 23, 2021
Version 4.4.0 has been released and includes updated recommendations and literature summary on neutralizing antibodies.
June 3, 2021
Version 4.3.0 has been released and contains a new recommendation on the use of remdesivir.
May 3, 2021
Version 4.2.1 has been released and contains endorsement from the Pediatric Infectious Diseases Society.
April 14, 2021
April 5, 2021
March 18, 2021
Version 4.1.1 has been released and contains a revision to the number of studies found for ivermectin.
March 5, 2021
Version 4.1.0 has been released and contains a new recommendation on the use of bamlanivimab with etesevimab among ambulatory patients.
February 22, 2021
Version 4.0.0 has been released and contains a revised recommendation on the use of tocilizumab.
February 18, 2021
Version 3.10.0 has been released and includes additional information on study eligibility for ivermectin.
February 10, 2021
Version 3.9.0 has been released and contains an updated literature review for tocilizumab.
February 5, 2021
Version 3.8.0 has been released and includes two new recommendations on the use of ivermectin.
February 3, 2021
Version 3.7.0 has been released and includes two new recommendations on the use of neutralizing monoclonal antibodies.
January 8, 2021
Version 3.6.0 has been realeased and includes new recommendations on the use of baricitinib and an updated literature review on hydroxychloroquine.
December 2, 2020
Version 3.5.1 has been released and includes endorsement from the Pediatric Infectious Diseases Society.
November 22, 2020
Version 3.5.0 has been released and includes revisions to the sections on lopinavir/ritonavir, tocilizumab, and remdesivir.
November 18, 2020
Version 3.4.0 has been released and contains a new recommendation on the use of bamlanivimab.
September 25, 2020
Version 3.3.0 has been released and contains revised and new recommendations for the use of dexamethasone and a revised recommendation against the routine use of tocilizumab.
September 21, 2020
Version 3.2.1 has been released and includes endorsement from the Pediatric Infectious Diseases Society.
September 15, 2020
Version 3.2.0 has been released and contains a new recommendation on the use of remdesivir in patients with more moderate disease.
September 4, 2020
Version 3.1.0 has been released and contains additional information on convalescent plasma as well new and updated narrative summaries of treatments undergoing evaluation.
August 28, 2020
Version 3.0.1 has been released and includes endorsement from the Pediatric Infectious Diseases Society.
August 20, 2020
Version 3.0.0 of the guideline has been released and contains revised recommendations on hydroxychloroquine and hydroxychloroquine plus azithromycin.
June 25, 2020
Version 2.1.0 of the guideline has been released and includes revised recommendations on corticosteroids.
June 22, 2020
Version 2.0.0 of the guideline has been released and contains:
- Revised recommendations on hydroxychloroquine and hydroxychloroquine plus azithromycin
- Revised recommendations for convalescent plasma for treatment of COVID-19
- New recommendations on the use of remdesivir
- New recommendations for famotidine (not addressed in versions 1.0.0-1.0.4)
April 21, 2020
Version 1.0.4 of the guideline has been released.
Overview of COVID-19 Treatment Guidelines (Summary Table)

Strengths of recommendation
Recommend (strong recommendation): Guideline panel is confident that the desirable effects of an intervention outweigh the undesirable effects. Most or all individuals will be best served by the recommended course of action.
Suggest (weak or conditional recommendation): Guideline panel after discussion concludes that the desirable effects probably outweigh undesirable effects, but appreciable uncertainty exists. Not all individuals will be best served by the recommended course of action and the caregiver needs to consider more carefully than usual the individual patient’s circumstances, preferences, and values.
Certainty of evidence
⨁⨁⨁⨁ high
⨁⨁⨁◯ moderate
⨁⨁◯◯ low
⨁◯◯◯ very low
See Figure 1 in the Executive Summary.
Abstract
Background: There are many pharmacologic therapies that are being used or considered for treatment of coronavirus disease 2019 (COVID-19), with rapidly changing efficacy and safety evidence from trials.
Objective: Develop evidence-based, rapid, living guidelines intended to support patients, clinicians, and other healthcare professionals in their decisions about treatment and management of patients with COVID-19.
Methods: In March 2020, the Infectious Diseases Society of America (IDSA) formed a multidisciplinary guideline panel of infectious disease clinicians, pharmacists, and methodologists with varied areas of expertise to regularly review the evidence and make recommendations about the treatment and management of persons with COVID-19. The process used a living guideline approach and followed a rapid recommendation development checklist. The panel prioritized questions and outcomes. A systematic review of the peer-reviewed and grey literature was conducted at regular intervals. The Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach was used to assess the certainty of evidence and make recommendations.
Results: Based on the most recent search conducted on May 31, 2022, the IDSA guideline panel has made 31 recommendations for the treatment and management of the following groups/populations: pre- and post-exposure prophylaxis, ambulatory with mild-to-moderate disease, hospitalized with mild-to-moderate, severe but not critical, and critical disease. As these are living guidelines, the most recent recommendations can be found online at: https://idsociety.org/COVID19guidelines.
Conclusions: At the inception of its work, the panel has expressed the overarching goal that patients be recruited into ongoing trials. Since then, many trials were done which provided much needed evidence for COVID-19 therapies. There still remain many unanswered questions as the pandemic evolved which we hope future trials can answer.
Keywords: coronavirus, SARS-CoV-2, COVID, COVID-19, pneumonia
Executive Summary and Background
Executive Summary
Coronavirus disease 2019 (COVID-19) is a pandemic with a rapidly increasing incidence of infections and deaths. Many pharmacologic therapies are being used or considered for treatment. Given the rapidity of emerging literature, the Infectious Diseases Society of America (IDSA) identified the need to develop living, frequently updated evidence-based guidelines to support patients, clinicians and other health-care professionals in their decisions about treatment and management of patients with COVID-19. Please refer to the IDSA website for the latest version of the guidelines: https://idsociety.org/COVID19guidelines.
Summarized below are the recommendations with comments related to the clinical practice guideline for the treatment and management of COVID-19. A detailed description of background, methods, evidence summary and rationale that support each recommendation, and research needs can be found online in the full text. In brief, per Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology, recommendations are labeled as “strong” or “conditional”. The word “recommend” indicates strong recommendations and “suggest” indicates conditional recommendations. In situations where promising interventions were judged to have insufficient evidence of benefit to support their use and with potential appreciable harms or costs, the expert panel recommended their use in the context of a clinical trial. These recommendations acknowledge the current “knowledge gap” and aim at avoiding premature favorable recommendations for potentially ineffective or harmful interventions.
Hydroxychloroquine/Chloroquine + Azithromycin
- Recommendation 1: Among patients with COVID-19, the IDSA guideline panel recommends against hydroxychloroquine. (Strong recommendation, Moderate certainty of evidence)
- Remark: Chloroquine is considered to be class equivalent to hydroxychloroquine.
- Recommendation 2: Among hospitalized patients with COVID-19, the IDSA guideline panel recommends against hydroxychloroquine plus azithromycin. (Strong recommendation, Low certainty of evidence)
- Remark: Chloroquine is considered to be class equivalent to hydroxychloroquine.
Hydroxychloroquine for Prophylaxis
- Recommendation 3: In persons exposed to COVID-19, the IDSA guideline panel recommends against hydroxychloroquine. (Strong recommendation, Moderate certainty of evidence)
Lopinavir/Ritonavir
- Recommendation 4: In persons exposed to COVID-19, the IDSA guideline panel recommends against post-exposure prophylaxis with lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
- Recommendation 5: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA guideline panel recommends against the use of lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
- Recommendation 6: Among hospitalized patients with COVID-19, the IDSA guideline panel recommends against the use of the combination lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
Glucocorticoids
- Recommendation 7: Among hospitalized critically ill patients* with COVID-19, the IDSA guideline panel recommends dexamethasone rather than no dexamethasone. (Strong recommendation, Moderate certainty of evidence)
- Remark: If dexamethasone is unavailable, equivalent total daily doses of alternative glucocorticoids may be used. Dexamethasone 6 mg IV or PO for 10 days (or until discharge) or equivalent glucocorticoid dose may be substituted if dexamethasone is unavailable. Equivalent total daily doses of alternative glucocorticoids to dexamethasone 6 mg daily are methylprednisolone 32 mg and prednisone 40 mg.
- Recommendation 8: Among hospitalized patients with severe**, but non-critical, COVID-19, the IDSA guideline panel suggests dexamethasone rather than no dexamethasone. (Conditional recommendation†, Moderate certainty of evidence)
- Remark: Dexamethasone 6 mg IV or PO for 10 days (or until discharge) or equivalent glucocorticoid dose may be substituted if dexamethasone is unavailable. Equivalent total daily doses of alternative glucocorticoids to dexamethasone 6 mg daily are methylprednisolone 32 mg and prednisone 40 mg.
- Recommendation 9: Among hospitalized patients with mild-to-moderate*** COVID-19 without hypoxemia requiring supplemental oxygen, the IDSA guideline panel suggests against the use of glucocorticoids. (Conditional recommendation††, Low certainty of evidence)
Severity definitions:
*Critical illness is defined as patients on mechanical ventilation and extracorporeal mechanical oxygenation (ECMO). Critical illness includes end organ dysfunction as is seen in sepsis/septic shock. In COVID-19, the most commonly reported form of end organ dysfunction is ARDS.
**Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
***Mild-to-moderate illness is defined as patient with a SpO2 >94% not requiring supplemental oxygen.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Inhaled Corticosteroids
- Recommendation 10: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA guideline panel suggests against inhaled corticosteroids. (Conditional recommendation††, Moderate certainty of evidence)
- Remark: Patients who are on inhaled corticosteroids for other indications may continue them.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Interleukin-6 Inhibitors
- Recommendation 11: Among hospitalized adults with progressive severe* or critical** COVID-19 who have elevated markers of systemic inflammation, the IDSA guideline panel suggests tocilizumab in addition to standard of care (i.e., steroids) rather than standard of care alone. (Conditional recommendation†, Low certainty of evidence)
- Remarks:
- Patients, particularly those who respond to steroids alone, who put a high value on avoiding possible adverse events of tocilizumab and a low value on the uncertain mortality reduction, would reasonably decline tocilizumab.
- In the largest trial on the treatment of tocilizumab, criterion for systemic inflammation was defined as CRP ≥75 mg/L.
- Remarks:
- Recommendation 12: When tocilizumab is not available, for patients who would otherwise qualify for tocilizumab, the IDSA guideline panel suggests sarilumab in addition to standard of care (i.e., steroids) rather than standard of care alone. (Conditional recommendation†, Very low certainty of evidence)
- Remark: Patients, particularly those who respond to steroids alone, who put a high value on avoiding possible adverse events of sarilumab and a low value on the uncertain mortality reduction, would reasonably decline sarilumab.
Severity definitions:
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
**Critical illness is defined as patients on mechanical ventilation and ECMO. Critical illness includes end organ dysfunction as is seen in sepsis/septic shock. In COVID-19, the most commonly reported form of end organ dysfunction is ARDS.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Convalescent Plasma
- Recommendation 13(UPDATED 2/22/2023): Among immunocompetent patients hospitalized with COVID-19, the IDSA guideline panel recommends against COVID-19 convalescent plasma. (Strong recommendation, Moderate certainty of evidence).
- Recommendation 14(NEW 2/22/2023): Among immunocompromised patients hospitalized with COVID-19, the IDSA guideline panel suggests against the routine use of COVID-19 convalescent plasma. (Conditional recommendation, very low certainty of evidence.
- Remark: Patients, particularly those who do not qualify for other treatments, who place a higher value on the uncertain mortality reduction and a lower value on the potential adverse effects of convalescent plasma would reasonably select convalescent plasma.
- Recommendation 15(UPDATED 2/22/2023): Among ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options*, the IDSA guideline panel suggests FDA-qualified high-titer COVID-19 convalescent plasma within 8 days of symptom onset rather than no high-titer COVID-19 convalescent plasma. (Conditional recommendation†, Low certainty of evidence)
- Remarks:
- In the United States, FDA emergency use authorization (EUA) only authorizes use in patients with immunosuppressive disease or receiving immunosuppressive treatment.
- Patients, particularly those who are not immunocompromised, who place a low value on the uncertain benefits (reduction in the need for mechanical ventilation, hospitalization, and death) and a high value on avoiding possible adverse events associated with convalescent plasma would reasonably decline convalescent plasma.
- Other options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir and three-day treatment with remdesivir Patient-specific factors (e.g., symptom duration, renal insufficiency or other contraindications, drug interactions) as well as logistical challenges, infusion capacity, and product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
- Remarks:
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Remdesivir
- Recommendation 16: Among patients (ambulatory or hospitalized) with mild-to-moderate COVID-19 at high risk for progression to severe disease, the IDSA guideline panel suggests remdesivir initiated within seven days of symptom onset rather than no remdesivir. (Conditional recommendation†, Low certainty of evidence)
- Remarks:
- Dosing for remdesivir in mild-to-moderate COVID-19 is 200 mg on day one followed by 100 mg on days two and three. Pediatric dosing is 5 mg/kg on day 1 and 2.5 mg/kg on subsequent days.
- Options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, three-day treatment with remdesivir, molnupiravir, and neutralizing monoclonal antibodies. Patient-specific factors (e.g., patient age, symptom duration, renal function, drug interactions), product availability, and institutional capacity and infrastructure should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
- Remarks:
- Recommendation 17: In patients on supplemental oxygen but not on mechanical ventilation or ECMO, the IDSA panel suggests treatment with five days of remdesivir rather than 10 days of remdesivir. (Conditional recommendation†, Low certainty of evidence)
- Recommendation 18a: In hospitalized patients with severe* COVID-19, the IDSA panel suggests remdesivir over no antiviral treatment. (Conditional recommendation†, Moderate certainty of evidence)
- Recommendation 18b: In patients with COVID-19 on invasive ventilation and/or ECMO, the IDSA panel suggests against the routine initiation of remdesivir (Conditional recommendation††, Very low certainty of evidence)
Severity definition:
*Severe illness is defined as patients with SpO2 ≤94% on room air.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Famotidine
- Recommendation 19: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA panel suggests against famotidine for the treatment of COVID-19 (Conditional recommendation††, Low certainty of evidence).
- Recommendation 20: Among hospitalized patients with severe* COVID-19, the IDSA panel suggests against famotidine for the treatment of COVID-19. (Conditional recommendation††, Low certainty of evidence)
Severity definition:
* Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Neutralizing Antibodies for Pre-Exposure Prophylaxis
As of 1/26/2023, based on CDC Nowcast data, fewer than 10% of circulating variants in the US are susceptible to tixagevimab/cilgavimab (Evusheld), the sole product that has been available for pre-exposure prophylaxis. Tixagevimab/cilgavimab is therefore no longer authorized for use in the US until further notice by FDA.
Neutralizing Antibodies for Post-Exposure Prophylaxis
The first two US FDA authorized anti-SARS-CoV-2 neutralizing antibody combinations, bamlanivimab/etesevimab and casirivimab/imdevimab, were found to be largely inactive against the Omicron BA.1 and BA.2 variants, rendering these products no longer useful for either treatment or post-exposure prophylaxis. As a result, Emergency Use Authorization was withdrawn by the US FDA for both bamlanivimab/etesevimab and casirivimab/imdevimab, leaving no available neutralizing antibody product for use in the United States for post-exposure prophylaxis.
Neutralizing Antibodies for Treatment
During 2022, multiple Omicron sub-variants with progressively greater in vitro reductions in susceptibility to multiple anti-SARS CoV-2 neutralizing antibodies emerged. On November 30, 2022, the US FDA withdrew Emergency Use Authorization for bebtelovimab, the one anti-SARS CoV-2 neutralizing antibody product that had retained in vitro activity against most previously circulating SARS-CoV-2 variants, leaving no available neutralizing antibody product in the United States for treatment of COVID-19.
Janus Kinase Inhibitors
- Recommendation 21: Among hospitalized adults with severe* COVID-19, the IDSA panel suggests baricitinib with corticosteroids rather than no baricitinib. (Conditional recommendation†, Moderate certainty of evidence)
- Remarks:
- Baricitinib 4 mg per day (or appropriate renal dosing) up to 14 days or until discharge from hospital.
- Baricitinib appears to demonstrate the most benefit in those with severe COVID-19 on high-flow oxygen/non-invasive ventilation at baseline.
- Limited additional data suggest a mortality reduction even among patients requiring mechanical ventilation.
- Remarks:
- Recommendation 22: Among hospitalized patients with severe* COVID-19 who cannot receive a corticosteroid (which is standard of care) because of a contraindication, the IDSA guideline panel suggests use of baricitinib with remdesivir rather than remdesivir alone. (Conditional recommendation†, Low certainty of evidence)
- Remark: Baricitinib 4 mg daily dose for 14 days or until hospital discharge. The benefits of baricitinib plus remdesivir for persons on mechanical ventilation are uncertain.
- Recommendation 23: Among hospitalized adults with severe** COVID-19 but not on non-invasive or invasive mechanical ventilation, the IDSA panel suggests tofacitinib rather than no tofacitinib. (Conditional recommendation†, Low certainty of evidence)
- Remarks:
- Tofacitinib appears to demonstrate the most benefit in those with severe COVID-19 on supplemental or high-flow oxygen.
- Patients treated with tofacitinib should be on at least prophylactic dose anticoagulant.
- Patients who receive tofacitinib should not receive tocilizumab or other IL-6 inhibitor for treatment of COVID-19.
- The STOP-COVID Trial did not include immunocompromised patients.
- Remarks:
Severity definitions:
* Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen, oxygen through a high-flow device, or non-invasive ventilation.
**Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen or oxygen through a high-flow device.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Ivermectin
- Recommendation 24: In hospitalized patients with COVID-19, the IDSA panel suggests against ivermectin. (Conditional recommendation††, Very low certainty of evidence)
- Recommendation 25: In ambulatory persons with COVID-19, the IDSA panel recommends against ivermectin. (Strong recommendation, Moderate certainty of evidence)
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Fluvoxamine
- Recommendation 26: Among ambulatory patients with COVID-19, the IDSA guideline panel recommends fluvoxamine only in the context of a clinical trial. (Knowledge gap)
Nirmatrelvir/Ritonavir
- Recommendation 27(UPDATED 4/12/2023): In ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease, the IDSA guideline panel suggests nirmatrelvir/ritonavir initiated within five days of symptom onset rather than no nirmatrelvir/ritonavir. (Conditional recommendation†, Low certainty of evidence)
- Remarks:
- Patients’ medications need to be screened for serious drug interactions
- Dosing based on renal function:
- Estimated glomerular filtration rate (eGFR) > 60 ml/min: 300 mg nirmatrelvir/100 ritonavir every 12 hours for five days
- eGFR ≤60 mL/min and ≥30 mL/min: 150 mg nirmatrelvir/100 mg ritonavir every 12 hours for five days
- eGFR <30 mL/min: not recommended
- Patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease admitted to the hospital may also receive nirmatrelvir/ritonavir
- Options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, remdesivir for a 3-day course, molnupiravir, and neutralizing monoclonal antibodies. Patient-specific factors (e.g., symptom duration, renal function, drug interactions) as well as product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
- Remarks:
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Molnupiravir
- Recommendation 28: In ambulatory patients (≥18 years) with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options*, the IDSA guideline panel suggests molnupiravir initiated within five days of symptom onset rather than no molnupiravir. (Conditional recommendation†, Low certainty of evidence)
*Other options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, three-day treatment with remdesivir, Patient-specific factors (e.g., symptom duration, renal function, drug interactions) as well as product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
- Remarks:
- Patients who will most likely benefit from antivirals are those with risk factors for progression to severe disease (e.g., elderly, those with high-risk comorbidities, incomplete vaccination status, or immunocompromised). Those without risk factors are less likely to benefit.
- Patients who put a higher value on the putative mutagenesis, adverse events, or reproductive concerns and a lower value on the uncertain benefits would reasonably decline molnupiravir.
- Patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease admitted to the hospital for reasons other than COVID-19 may also receive molnupiravir.
- Molnupiravir is not authorized under the FDA EUA for use in patients <18 years because it may affect bone and cartilage growth.
- Molnupiravir is not recommended under the FDA EUA for use during pregnancy.
- Molnupiravir is not authorized under the FDA EUA for pre-exposure or post-exposure prevention of COVID-19 or for initiation of treatment in patients hospitalized due to COVID-19 because benefit of treatment has not been observed in individuals when treatment is started after hospitalization due to COVID-19.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Colchicine
- Recommendation 29: In hospitalized patients with COVID-19, the IDSA panel recommends against colchicine for treatment of COVID-19. (Strong recommendation, Moderate certainty of evidence)
- Recommendation 30: In ambulatory persons with COVID-19, the IDSA panel suggests against colchicine for treatment of COVID-19. (Conditional recommendation††, Moderate certainty of evidence)
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Anakinra
- Recommendation 31(NEW 5/4/2023): In hospitalized patients with severe* COVID-19, the IDSA guideline panel suggests against the routine use of anakinra. (Conditional recommendation, Low certainty of evidence)
Severity definitions:
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
At the inception of its work, the panel expressed the overarching goal that patients be recruited into ongoing trials, which would provide much needed evidence on the efficacy and safety of various therapies for COVID-19. Since then, many trials were done which provided much needed evidence for COVID-19 therapies. There still remain many unanswered questions as the pandemic evolved which we hope future trials can answer. The panel has determined that when an explicit trade-off between highly uncertain benefits and known putative harms of these therapeutic agents were considered, a net positive benefit was not reached and could possibly be negative (risk of excess harm). The panel acknowledges that enrolling patients in randomized controlled trials (RCTs) might not be feasible for many frontline providers due to limited access and infrastructure. Should lack of access to clinical trials exist, we encourage setting up local or collaborative registries to systematically evaluate the efficacy and safety of drugs to contribute to the knowledge base. Each clinician can play a role in advancing our understanding of this disease through a local registry or other data collection efforts.
Background
The first cases of COVID-19 were reported from Wuhan, China in early December 2019 [1], now known to be caused by a novel beta-coronavirus, named as Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Within a span of months, COVID-19 has become pandemic due to its transmissibility, spreading across continents with the number of cases and deaths rising daily [2]. The emergence of new variants as the pandemic evolved has added more challenges to the prevention and treatment of COVID-19. Although most infected individuals exhibit a mild illness (80%+), 14% have serious and 5% have critical illness. Approximately 10% will require hospital admission due to COVID-19 pneumonia, of which approximately 10% will require intensive care, including invasive ventilation due to acute respiratory distress syndrome (ARDS) [3]. While mortality appears to be more common in older individuals and those with comorbidities, such as chronic lung disease, cardiovascular disease, hypertension and diabetes, young people with no comorbidities also appear to be at risk for critical illness including multi-organ failure and death.
There has been an expanding number of studies rapidly published online and in academic journals; however, some of these may be of limited quality and are pre-published without sufficient peer-review. Critical appraisal of the existing studies is needed to determine if the existing evidence is sufficient to support currently proposed management strategies.
Given the rapid global spread of SARS-CoV-2 and the difficulty for the overburdened front-line providers and policymakers to stay up to date on emerging literature, IDSA has recognized the necessity of developing a rapid guideline for the treatment of COVID-19. The guideline panel is using a methodologically rigorous process for evaluating the best available evidence and providing treatment recommendations. These guidelines will be frequently updated as substantive literature becomes available and are accessible on an easy to navigate web and device interface at http://www.idsociety.org/covid19guidelines.
There continue to be several ongoing trials evaluating therapeutic agents for the treatment of COVID-19. As data becomes available from these trials and if there is a preponderance of evidence to suggest the use of a therapeutic agent even in the context of clinical trials is no longer warranted it will be removed from future updates of the guideline (and the removal will be noted in the updated guidelines). If there is emerging evidence on the efficacy or safety of a therapeutic agent not mentioned in the current version of the guideline it will be included in future updates of the guideline.
These recommendations are intended to inform patients, clinicians, and other health professionals by providing the latest available evidence.
Methods and Search Results
This guideline was developed in two stages. First, an initial rapid systematic review was conducted to inform the first iteration of the guideline. Second, while maintaining a current evidence based, the guideline scope expanded to update existing recommendations and include additional therapies, as needed, using a living guideline approach. Given the need for continued urgent responses to this major public health crisis, the methodological approach follows the Guidelines International Network/McMaster checklist for the development of rapid recommendations [4].
Panel Composition
The initial guideline panel assembled in March 2020 was composed of nine members including infectious diseases specialists as well as experts in public health as well as other front-line clinicians, specializing in pharmacology, pediatrics, medical microbiology, preventive care, critical care, hepatology, nephrology and gastroenterology. Organizational representatives were included from the Society for Healthcare Epidemiology of America (SHEA) and the Pediatric Infectious Diseases Society (PIDS). In May 2020, an additional panel member was included as a representative from the Society of Infectious Diseases Pharmacists (SIDP). One member rotated off the panel in March of 2022 and replaced by a Pediatric ID specialist and an adult ID specialist with expertise in antiviral drug resistance testing. The Evidence Foundation provided technical support and guideline methodologists for the development of this guideline.
Disclosure and Management of Potential Conflicts of Interest
All members of the expert panel complied with the COI process for reviewing and managing conflicts of interest, which requires disclosure of any financial, intellectual, or other interest that might be construed as constituting an actual, potential, or apparent conflict, regardless of relevancy to the guideline topic. The assessment of disclosed relationships for possible COI is based on the relative weight of the financial relationship (i.e., monetary amount) and the relevance of the relationship (i.e., the degree to which an association might reasonably be interpreted by an independent observer as related to the topic or recommendation of consideration). The COI review group has ensured that the majority of the panel and chair is without potential relevant (related to the topic) conflicts for the duration of their term on the panel. The chair and all members of the technical team have been determined to be unconflicted.
Question Generation
Clinical questions included in this guideline were developed into a PICO format (Population, Intervention, Comparison, Outcomes) [5] and prioritized according to available evidence that met the minimum acceptable criteria (i.e., the body of evidence reported on at least a case-series design, case reports were excluded). Panel members prioritized patient-important outcomes such as mortality, hospitalization, development of severe disease (e.g., need for non-invasive or invasive ventilation) and clinical improvement (such as disease-oriented outcomes inferred by radiological findings or virologic cure), and severe adverse events leading to treatment discontinuation. Serious adverse events are death, life threatening reactions, those that require hospitalization, result in disability or permanent damage or require an intervention to prevent permanent impairment [6]. Additional drug specific harms were evaluated when clinically relevant, including possible drug-drug reactions, if applicable.
Critical and important outcomes for decision-making varied across populations/groups. For example, among hospitalized patients (at any disease severity), critical outcomes included mortality, need for invasive mechanical ventilation, duration of hospitalization, failure of clinical improvement, adverse events, and serious adverse events. Among ambulatory populations with COVID-19 infection, the outcome of hospitalization replaced duration of hospitalization. Among persons receiving pre- or post-exposure prophylaxis, outcomes included measures of symptomatic COVID-19 infection.
Search Strategy
The National Institute for Health and Care Excellence (NICE) highly-sensitive search was reviewed by the methodologist in consultation with the technical team information specialist and was determined to have high sensitivity [7]. An additional term, COVID, was added to the search strategy used, in addition to the treatment terms identified in the PICO questions (Supplementary Table s1). Per living guideline approach, monthly searches are conducted in Ovid Medline and Embase, building on the literature searched from 2019. This document reflect literature searched through May 31, 2022. Horizon scans have been performed regularly during the evidence assessment and recommendation process to locate additional grey literature, including manuscript pre-prints. Reference lists and literature suggested by panelists were reviewed for inclusion. No restrictions were placed on language or study type.
Screening and Study Selection
Two reviewers independently screened titles and abstracts, as well as eligible full-text studies. Eligible studies reported on persons with confirmed COVID-19 and compared the active intervention against no active intervention (e.g., standard of care or other treatment equally distributed across both the intervention and comparison arm). For questions on pre- or post-exposure prophylaxis, persons at baseline could not have reported COVID-19 infection. When acceptable RCTs of effectiveness were found, no additional non-randomized studies or non-comparative evidence (i.e., single-arm case series) were sought. Evidence from single arm studies reporting on non-comparative rates of outcomes of interest were included if a historical control event rate could be estimated from the literature. Conflicts were resolved through discussion or with a third reviewer.
Data Collection and Analysis
Reviewers extracted relevant information into a standardized data extraction form, including: study characteristics, study design, participant characteristics, details of the intervention and comparison, outcomes reported and funding source. We extracted number of events and total sample to calculate a risk ratio and corresponding 95% confidence interval (CI) for dichotomous outcomes. For continuous outcomes, either a mean and standard deviation or a standard mean difference were calculated. Where applicable, data were pooled using random effects model (fixed effects model for two or fewer trials or pooling of rates) and presented in a forest plot using RevMan [8].
Risk of Bias and Certainty of Evidence
Risk of bias was assessed using the Cochrane Risk of Bias Tool for RCTs and the Risk of Bias Instrument for Non-randomized Studies – of Interventions (ROBINS-I) [9, 10]. The certainty of evidence was assessed using the GRADE approach [11]. Within GRADE, the body of evidence across each outcome is assessed for domains that may reduce or increase one’s certainty in the evidence. Factors that may reduce one’s certainty include risk of bias (study limitations), inconsistency (unexplained heterogeneity across study findings), indirectness (applicability or generalizability to the research question), imprecision (the confidence in the estimate of an effect to support a particular decision) or publication bias (selective publication of studies). One’s certainty in the evidence may be strengthened if the following considerations are present: large or very large magnitude of effect, evidence of a dose-response gradient, or opposing residual confounding. GRADE summary of findings tables were developed in GRADEpro Guideline Development Tool [12].
The outcomes informing decision-making for specific treatments may change to reflect the availability of higher-quality direct evidence for critical clinical outcomes. For example, at the time of the first guideline, clinical improvement outcomes (e.g., need for mechanical ventilation) were not reported, only the results of radiographic findings. However, with the recent publication of RCTs and non-randomized studies reporting on direct measures of clinical improvement, results of radiographic studies were deemed to be less critical for decision making.
Evidence to Recommendations
The panel considered core elements of the GRADE evidence in the decision process, including Certainty of evidence and balance between desirable and undesirable effects. Additional domains were acknowledged where applicable (feasibility, resource use, acceptability). For all recommendations, the expert panelists reached consensus. Voting rules were agreed on prior to the panel meetings for situations when consensus could not be reached. If the panel is deciding because a strong or a conditional recommendation (based on moderate or high certainty evidence) in the same direction, 80% of the panel must vote for a strong recommendation. In situations of uncertainty between the desirable and undesirable consequences (typically based on low or very low certainty evidence), when the panel is deciding between a conditional recommendation or no recommendation, 50% of the panel must vote for the same option with less than 20% voting for the alternative option.
As per GRADE methodology, recommendations are labeled as “strong” or “conditional”. The words “we recommend” indicate strong recommendations and “we suggest” indicate conditional recommendations. Figure 1 provides the suggested interpretation of strong and weak recommendations for patients, clinicians, and healthcare policymakers. For recommendations where the comparators are not formally stated, the comparison of interest is implicitly referred to as “not using the intervention”. These recommendations acknowledge the current “knowledge gap” and aim at avoiding premature favorable recommendations for their use and to avoid encouraging the rapid diffusion of potentially ineffective or harmful interventions. Detailed suggestions about the specific research questions that should be addressed are found in the table (see Supplementary Table s2).
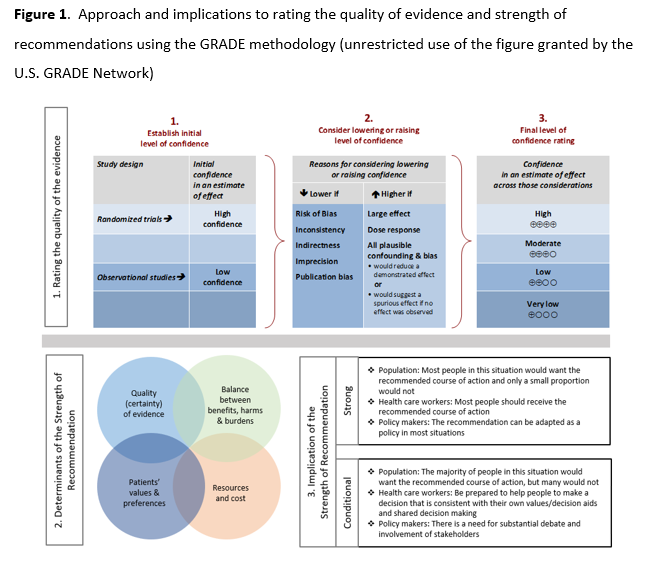
Review Process
This guideline has been rapidly reviewed and approved by the IDSA Board of Directors Executive Committee external to the guideline development panel. SHEA, PIDS, and SIDP have reviewed and provided endorsement of its contents.
Updating Process and Terminology
As detailed in the methods section, the living guideline is supported by monthly screening of the literature. The impetus for updating a current recommendation is based on the identification of peer-reviewed or publicly-available, grey literature reporting data for at least one critical outcome that would likely have an impact on the recommendations. This could reflect new information on a critical outcome that previously had no included evidence, changes to the absolute effect of a critical outcome (magnitude or precision), or changes to the certainty of a critical outcome. In such situations, the entire expert panel is reconvened to review the evidence and put forward a proposal for a change in the recommendation.
Changes to these guidelines falls into one of three categories: update, amendment, or retirement. An update involves a search for new studies, and if any new studies are found, they will be critically appraised and the pertinent section will be removed and replaced with the updated section. An amendment involves a change or correction to the document without any search for new studies and their appraisal. It will also involve changes made to clarify or explain a section based on “living” feedback from the readers. Due to lack of continued relevancy of a treatment option, the guideline panel may choose to retire a section. While the retired section will not appear in the manuscript, all sections with accompanying dates will be available on the IDSA website.
Guideline revisions may result in major, minor, or “patch” version changes, defined as follows:
- Major version (e.g., 1.0.0): Synonymous with a newly published version in the journal. This is usually called a "breaking version", i.e. prior recommendations may not be valid anymore.
- Minor version (e.g., 1.1.0): Includes new information, maybe even added PICOs, but not a breaking version, i.e., existing recommendations are still valid, although new recommendations may be available.
- Patch version (e.g., 1.0.1): Small changes, i.e., typos, adding words, removing words, but there are no material changes to the document or changes in recommendations.
Results
Systematic review and horizon scan of the literature identified 68,968 references of which 147 informed the evidence base for these recommendations (Supplementary Figure s1). Characteristics of the included studies can be found in the supplementary materials.
Recommendations 1-2: Hydroxychloroquine and Hydroxychloroquine + azithromycin
Section last reviewed and updated 12/23/2020
Last literature search conducted 12/14/2020
Recommendation 1: Among hospitalized patients with COVID-19, the IDSA guideline panel recommends against hydroxychloroquine*. (Strong recommendation, Moderate certainty of evidence)
- Remark: Chloroquine is considered to be class equivalent to hydroxychloroquine.
Recommendation 2: Among hospitalized patients with COVID-19, the IDSA guideline panel recommends against hydroxychloroquine* plus azithromycin. (Strong recommendation, Low certainty of evidence)
- Remark: Chloroquine is considered to be class equivalent to hydroxychloroquine.
Why are hydroxychloroquine and hydroxychloroquine plus azithromycin considered for treatment?
Hydroxychloroquine (HCQ) and chloroquine are 4-aminoquinoline drugs developed in the mid-20th century for the treatment of malaria [13]. Hydroxychloroquine differs from chloroquine only in the addition of a hydroxyl group and is associated with a lower incidence of adverse effects with chronic use [13]. Both drugs have been used in the treatment of autoimmune diseases because of their immunomodulatory effects on several cytokines, including interleukin-1 (IL-1) and IL-6 [13]. There is some evidence that these drugs also have antiviral properties against many different viruses, including the coronaviruses [14, 15]. They have demonstrated in vitro activity against SARS-CoV-2, which range considerably between studies, but are generally within the range of predicted achievable tissue concentrations [14, 16-18]. The in vitro activity, the extensive use for other conditions, and widespread availability of generic versions of the drug made it an attractive option for treatment of COVID-19. Interest in combinations of HCQ with azithromycin (AZ) began when investigators in a small, uncontrolled study of hydroxychloroquine use for COVID-19 noticed a higher frequency of patients achieving virologic response in the six subjects who received AZ to prevent bacterial infection [19]. Azithromycin, widely utilized as an antibacterial agent, has also been shown to have in vitro antiviral activity against a variety of ribonucleic acid viruses [20-22]. While the exact mechanism of antiviral activity is unknown, possibilities include inhibiting endocytosis and limiting viral replication [23] and the induction of interferon [22, 24]. Macrolides have also been shown to have anti-inflammatory activity [25, 26].
Summary of the evidence
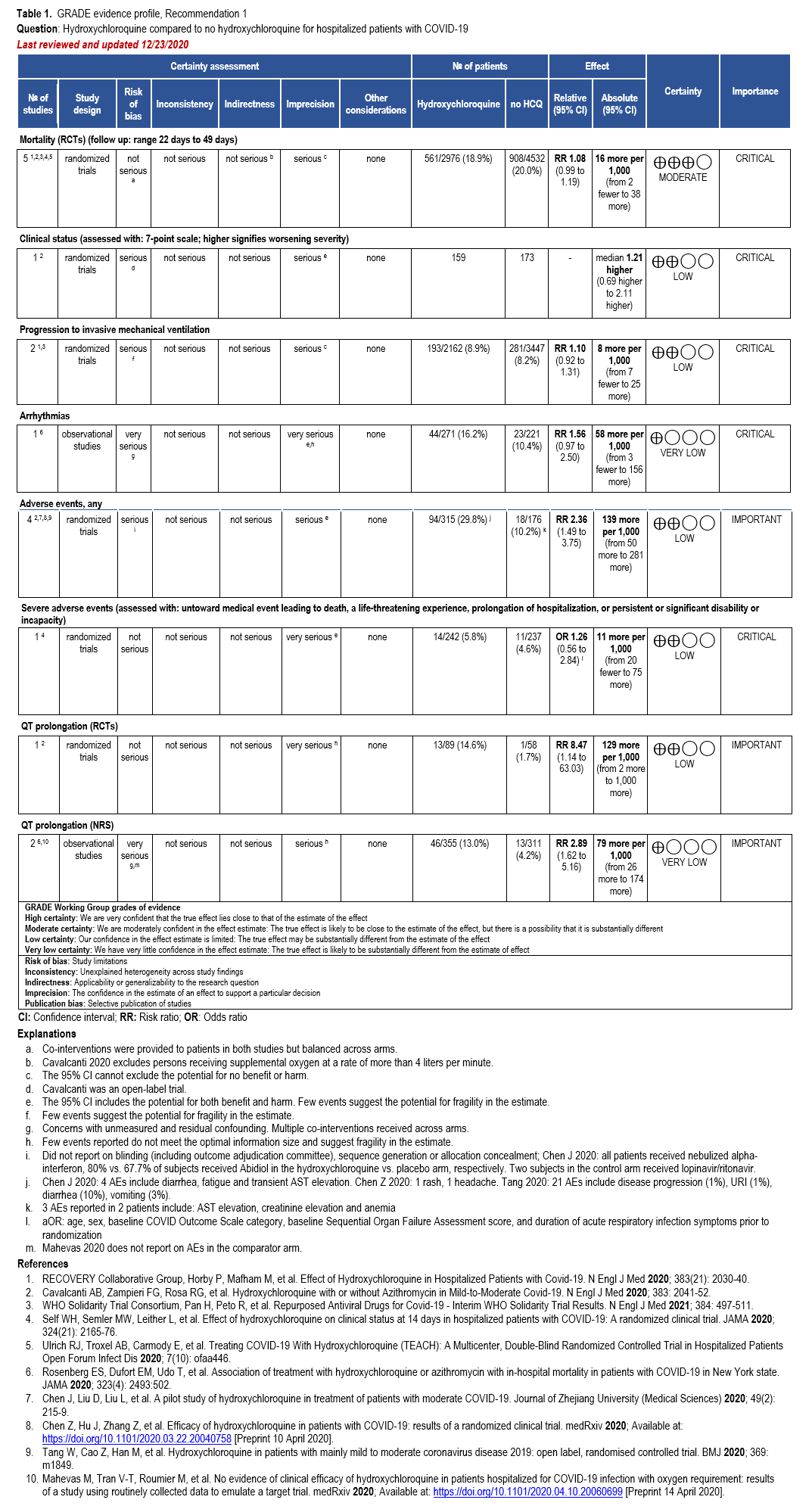
Our search identified eight RCTs and seven comparative cohort studies of hospitalized patients with confirmed COVID-19 treated with HCQ with reported mortality, clinical progression or clinical improvement, and adverse events outcomes [27-41] (Supplementary Table s3a) (Table 1).
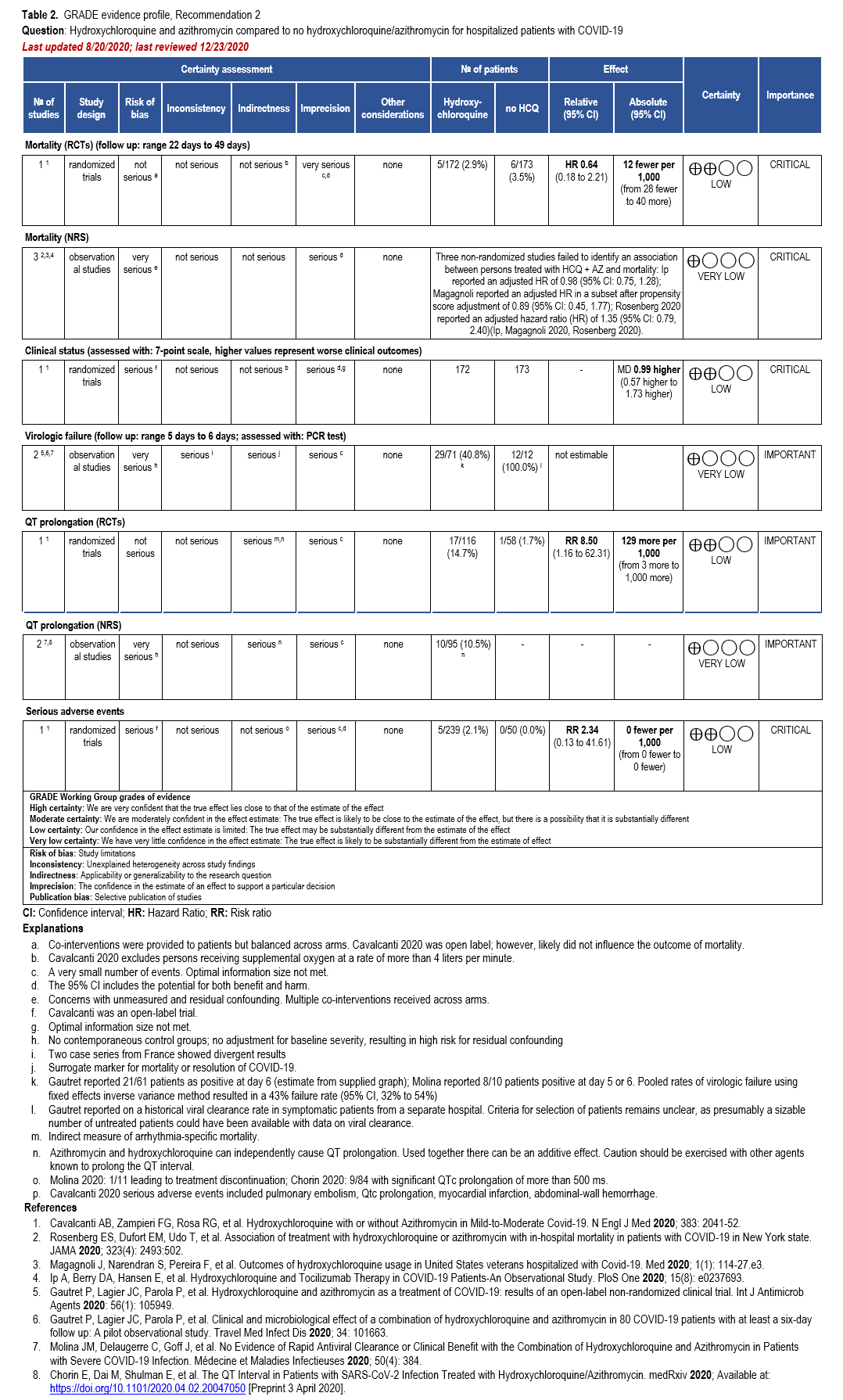
In addition, we identified two RCTs, four comparative cohort studies, one case-control study, and three single-arm studies reporting adjusted analyses of hospitalized patients with confirmed COVID-19 treated with HCQ plus AZ with reported mortality, failure of virologic clearance (assessed with polymerase chain reaction [PCR] test), clinical improvement, and adverse events (i.e., significant QT prolongation leading to treatment discontinuation) [19, 27, 28, 37, 39, 41-45] (Supplementary Table s3b) (Table 2).
Benefits
Hydroxychloroquine
Five RCTs showed a trend toward mortality among patients with COVID-19 treated with HCQ compared to those who were not (relative risk [RR]: 1.08; 95% confidence interval [CI]: 0.99, 1.19, Moderate certainty in the evidence) (Table 1) [28, 29, 33].
Hydroxychloroquine + Azithromycin
One RCT could not exclude the risk of in-hospital mortality among patients treated with HCQ+AZ compared to those not receiving HCQ or HCQ+AZ (hazard ratio [HR]: 0.64; 95% CI: 0.18, 2.21; Low certainty of evidence [CoE]) [28]. Three non-randomized studies failed to identify an association between treatment with HCQ+AZ and mortality: Ip reported an adjusted HR of 0.98 (95% CI: 0.75, 1.28); Magagnoli reported an adjusted HR in a subset after propensity score adjustment of 0.89 (95% CI: 0.45, 1.77); Rosenberg 2020 reported an adjusted HR of 1.35 (95% CI: 0.79, 2.40) [37, 39, 41]. As stated in the HCQ section, one non-randomized study reported a reduction in mortality among patients receiving HCQ+AZ (HR: 0.29; 95% CI: 0.22, 0.40); however, it failed to adjust for the critical confounder of disease severity and imbalances in steroid use [27]. As described in the HCQ section, similar methodologic concerns exist among patients allocated to HCQ+AZ in the Arshad study, leading to several sources of bias in interpreting their favorable results.
Harms
Hydroxychloroquine
One RCT reported that persons treated with HCQ experienced a longer time until hospital discharge (median 16 days compared with 13 days) and lower probability of being discharged alive within the 28-day study period (rate ratio: 0.92; 95% CI: 0.85, 0.99) [29]. In addition, persons treated with HCQ who were not on mechanical ventilation at baseline were more likely to be placed on mechanical ventilation during follow up (rate ratio: 1.10; 95% CI: 0.92, 1.31; Low CoE) [29, 32]. Across the body of evidence from four RCTs, treatment with HCQ may increase the risk of experiencing adverse events (RR: 2.36; 95% CI: 1.49, 3.75; Low CoE) and severe adverse events (adjusted odds ratio: 1.26; 95% CI: 0.56, 2.84; Low CoE) [28, 30, 31, 35]. One RCT and two non-randomized studies suggest increased risk of QT prolongation among patients treated with HCQ compared to those not receiving HCQ (RR: 8.47; 95% CI: 1.14, 63.03; Low CoE and RR: 2.89; 95% CI: 1.62, 5.16; Very low CoE, respectively) [28, 38, 39]. In addition, Rosenberg 2020 reported 16% of patients in the HCQ arm experienced arrhythmias compared with 10% in the non-HCQ arm (RR: 1.56; 95% CI: 0.97, 2.50; Very low CoE).
Gastrointestinal side effects occurred in 7% of patients in a prospective cohort study in 224 COVID-19 uninfected patients with systemic lupus erythematosus (SLE) who received either chloroquine or hydroxychloroquine for routine care [46].
While the 4-aminoquinolines, chloroquine and HCQ, have not been demonstrated to cause hemolysis in people with glucose-6-phosphate dehydrogenase (G6PD) deficiency [47, 48], case reports of hemolysis have emerged when these agents have been used for the treatment of COVID-19 [49-51]. It is possible that infection with SARS-CoV-2 may trigger hemolysis in G6PD deficient individuals in the absence of a 4-aminoquinolone. Caution should be exercised in administering these agents to G6PD deficient individuals with COVID-19, particularly if used for extended durations.
Renal clearance accounts for 15-25% of total clearance of HCQ; however, dose adjustments are not recommended with kidney dysfunction. Chloroquine and HCQ are metabolized by cytochrome P450 isoenzymes 2C8, 2D6, and 3A4 [52]. Therefore, inhibitors and inducers of these enzymes may result in altered pharmacokinetics of these agents.
Hydroxychloroquine + Azithromycin
One RCT suggests increased risk of QT prolongation among patients treated with HCQ+AZ compared to those not receiving HCQ (RR: 8.50; 95% CI: 1.16, 62.31; Low CoE) [28]. Two studies described significant QT prolongation in 10 of 95 patients treated with HCQ+AZ, illustrating the high risk for clinically relevant arrhythmias with this treatment [43, 45]. In addition, several case reports of QT prolongation related to HCQ have also been published [53-56]. A case-control study of persons with COVID-19 treated with HCQ+AZ compared to healthy, untreated controls reported higher values of minimum (415 vs. 376 ms), mean (453 vs. 407 ms) and maximum QTc-interval (533 vs. 452 ms) among COVID-19 cases (n=22) compared to controls (n=34) [42].
Additional case reports have cited the risk of a prolonged QT prolongation, torsades de pointes, and ventricular tachycardia in patients without COVID-19 receiving AZ alone. In a large cohort study, patients taking a five-day course of AZ had an increased risk of sudden cardiac death with a HR of 2.71 (1.58-4.64) vs. 0.85 (0.45-1.60), compared to patients receiving either no antibiotic or amoxicillin, respectively [57]. Given the cumulative effect on cardiac conduction seen with HCQ and AZ, if this combination was used, baseline and follow-up electrocardiogram (ECG) monitoring would be indicated, as well as careful surveillance for other concomitant medications known to prolong the QT interval.
Azithromycin has a low risk for cytochrome P450 interactions [58]; however, additional pharmacologic adverse events including gastrointestinal effects and QT prolongation need to be carefully considered, particularly in the outpatient setting where frequent ECG monitoring is not feasible.
Providers are encouraged to visit resources such as https://www.covid19-druginteractions.org/ to aid in the evaluation and management of drug interactions with current and emerging investigational agents for COVID-19.
Other considerations
The panel agreed that the overall certainty of evidence against treatment with HCQ was moderate due to concerns with imprecision around the risk for a trend towards harms from increased mortality. When considering the addition of AZ, the overall certainty of the evidence was low; however, the panel recognized even greater concern with the toxicity. In addition, based on the moderate certainty of increased QT prolongation, the panel determined that this demonstrated certain harm with uncertain benefit; therefore, the panel made a strong recommendation against HCQ+AZ.
Conclusions and research needs for this recommendation
The guideline panel recommends against the use of either HCQ alone or in combination with AZ in the hospital setting as higher certainty benefits (e.g., mortality reduction) are now highly unlikely even if additional high quality RCTs would become available.
This recommendation does not address the use of azithromycin for secondary bacterial pneumonia in patients with COVID-19 (Supplementary Table s2).
Supplementary Information
Study characteristics:
- Table s3a. Hydroxychloroquine versus no hydroxychloroquine
- Table s3b. Hydroxychloroquine/azithromycin versus no hydroxychloroquine/azithromycin
Forest plots:
- Figure s2a. Outcome of mortality point estimate demonstrates increased risk with hydroxychloroquine treatment
Figure s2b. Outcome of progression to mechanical ventilation demonstrates increased risk with HCQ treatment - Figure s2c. Outcome of adverse events demonstrates increased risk with hydroxychloroquine treatment
- Figure s2d. Outcome of QT prolongation demonstrates increased risk with hydroxychloroquine treatment
Risk of bias:
- Table s4a. Randomized controlled studies (hydroxychloroquine ± azithromycin vs. no hydroxychloroquine ± azithromycin)
- Table s4b. Non-randomized studies (hydroxychloroquine ± azithromycin vs. no hydroxychloroquine ± azithromycin)
Recommendation 3: Hydroxychloriquine as post-exposure prophylaxis
Section last reviewed and updated 9/23/2021
Last literature search conducted 9/21/2021
Recommendation 3: In persons exposed to COVID-19, the IDSA guideline panel recommends against hydroxychloroquine. (Strong recommendation, Low certainty of evidence)
Why is hydroxychloroquine considered for post-exposure prophylaxis?
There is some evidence that HCQ has antiviral properties against many different viruses, including the coronaviruses [14, 15]. It has demonstrated in vitro activity against SARS-CoV-2, which ranges considerably between studies, but is generally within the range of predicted achievable tissue concentrations [14, 16-18]. The in vitro activity, the extensive use for other conditions, and widespread availability of generic versions of the drug made it an attractive option for treatment and prophylaxis of COVID-19; however, at this point, HCQ has not been identified as effective for treatment of COVID-19.
Summary of the evidence
Our search identified three RCTs that reported on HCQ post-exposure prophylaxis of contacts of those diagnosed with SARS-CoV-2 infection [59-61]. Patients in these studies were randomized to HCQ or placebo or no additional treatment. All three studies evaluated for the presence of SARS-CoV-2 at day 14, two of the studies required a positive test for SARS-CoV-2, while one allowed symptoms suggestive of COVID-19 to meet the outcome when a test was not completed. Additional outcomes included hospitalization, mortality, and serious adverse events.
Benefits
Outpatients
Hydroxychloroquine appears to have trivial or no effect on the development of symptomatic SARS-CoV-2 infection at day 14 compared to no HCQ (RR: 0.95; 95% CI: 0.77, 1.16; moderate CoE). In addition, HCQ showed trivial or no effect on the rate of hospitalization (RR: 1.00; 95% CI: 0.47, 2.12; three fewer to seven more hospitalizations in 1,000; low CoE) or mortality (RR: 0.45; 95% CI: 0.16, 1.28; five fewer to two more deaths in 1,000; low CoE).
Harms
There was no difference in serious adverse events in the HCQ rather than no HCQ for post-exposure prophylaxis (RR: 0.91; 95% CI: 0.47, 1.76; low CoE). Additional side effects and harms of HCQ (e.g., QT prolongation, arrhythmias, gastrointestinal effects) have been summarized in recommendation 1 (HCQ for treatment of hospitalized persons with COVID-19).
Other considerations
The panel made an explicit decision that:
- The primary outcome driving the decision for any post-exposure prophylaxis is the ability to prevent infection
- When the evidence demonstrates a very low likelihood of effective post-exposure prophylaxis, other outcomes become secondary
- When healthy persons are considered for preventive medications (such as would occur in post-exposure settings), a higher threshold for benefits is required and (even putative) harms become more important
The panel agreed that the overall certainty of the evidence against prophylaxis treatment with HCQ was moderate (failure to prevent infection) due to concerns with imprecision. The panel balanced the lack of clear benefit with the increased risk of harms from the body of evidence reported in the treatment section, in addition to the side effects reported in the trials to make a strong recommendation.
Conclusions and research needs for this recommendation
The guideline panel recommended against the use of HCQ as post-exposure prophylactic treatment for persons exposed to COVID-19.
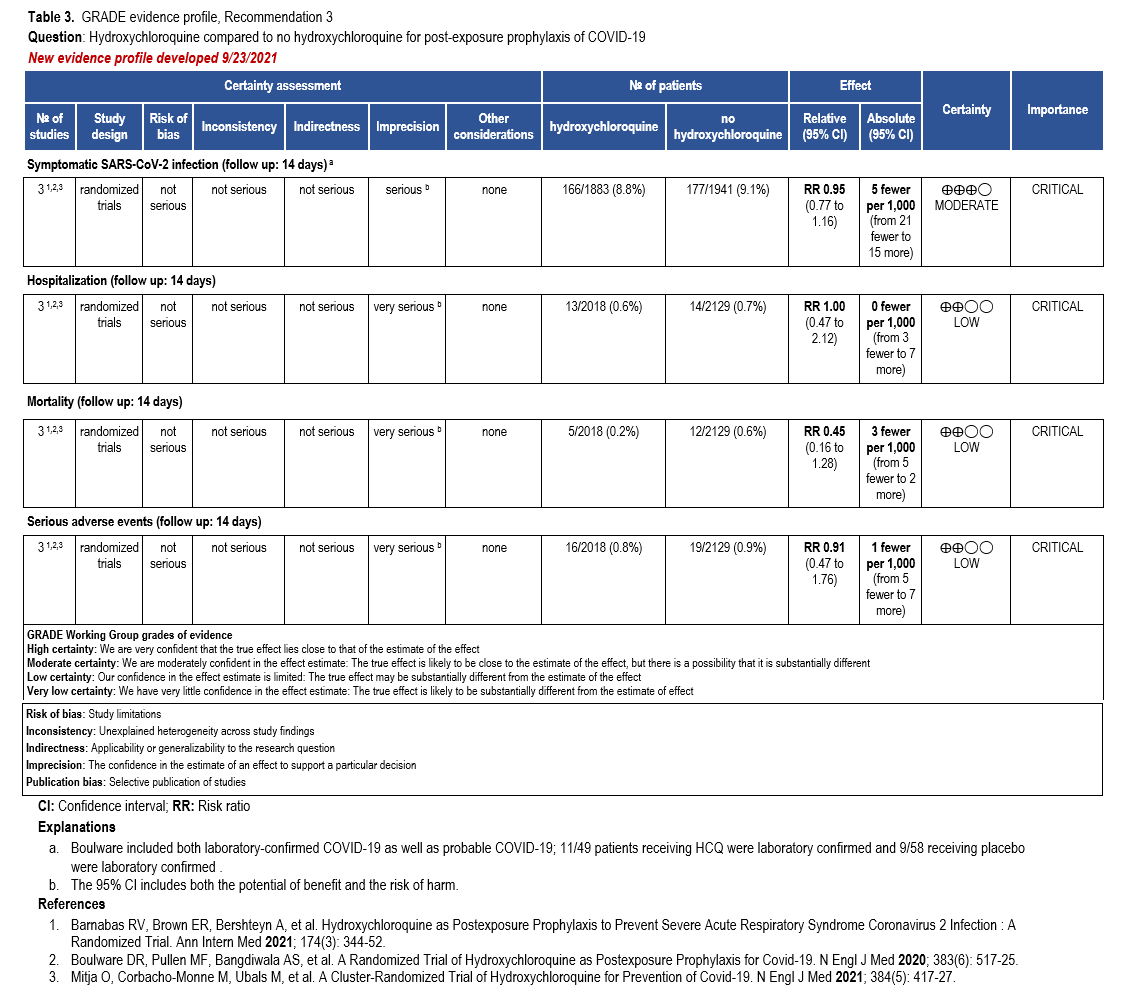
Supplementary Information
Study characteristics:
Forest plots:
- Figure s3a. Outcome of SARS-CoV-2 infection at 14 days for post-exposure hydroxychloroquine vs. no hydroxychloroquine for persons exposed to COVID-19
- Figure s3b. Outcome of hospitalization at 14 days for post-exposure hydroxychloroquine vs. no hydroxychloroquine for persons exposed to COVID-19
- Figure s3c. Outcome of mortality at 14 days for post-exposure hydroxychloroquine vs. no hydroxychloroquine for persons exposed to COVID-19
- Figure s3d. Outcome of serious adverse events at 14 days for post-exposure hydroxychloroquine vs. no hydroxychloroquine for persons exposed to COVID-19
Risk of bias:
Recommendations 4-6: Lopinavir/ritonavir
Section last reviewed and updated 2/16/2022
Last literature search conducted 1/31/2022
Recommendation 4: In persons exposed to COVID-19, the IDSA guideline panel recommends against post-exposure prophylaxis with lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
Recommendation 5: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA guideline panel recommends against the use of lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
Recommendation 6: Among hospitalized patients with COVID-19, the IDSA guideline panel recommends against the use of the combination lopinavir/ritonavir. (Strong recommendation, Moderate certainty of evidence)
Why is lopinavir plus ritonavir considered for treatment?
Lopinavir/ritonavir is a protease inhibitor that was U.S. Food and Drug Administration (FDA)-approved for the treatment of HIV in September 2000. Ritonavir is added to the combination as a pharmacokinetic enhancer due to its strong inhibition of cytochrome P450 3A4, a metabolic pathway for lopinavir metabolism. Lopinavir/ritonavir demonstrated in vitro inhibition of SARS-CoV-1 and MERS-CoV replication [62-64]. A trial of lopinavir/ritonavir and ribavirin versus historical controls in SARS-CoV-1 patients, showed a reduced rate of acute respiratory distress syndrome and mortality in those receiving lopinavir/ritonavir. This study had limitations including a control group from early in the outbreak when management strategies likely differed significantly [65]. During the MERS outbreak, case reports cited efficacy of lopinavir/ritonavir with interferon in the management of MERS patients [66, 67]. During the early phase of COVID-19, triple combination of interferon beta-1b, lopinavir/ritonavir, and ribavirin shortened the duration of viral shedding and hospital stay in patients with mild-to-moderate COVID-19 in an open-label, randomized, phase II trial [68].
Summary of the evidence
One RCT reported on post-exposure prophylaxis with combination lopinavir/ritonavir or placebo for ambulatory persons exposed to COVID-19 [69]. During the follow up period of 21 days, the investigators reported on symptomatic SARS-CoV-2 infection (COVID) either independent of baseline PCR/serology or among those who had a negative PCR test/serology at baseline.
One RCT reported on treatment with combination lopinavir/ritonavir or placebo for ambulatory patients with mild-to-moderate COVID-19 [70]. During the follow up of 90 days, COVID-19-related hospitalizations as well as mortality were recorded.
Three RCTs reported on treatment with combination lopinavir/ritonavir or placebo for hospitalized patients with COVID-19 [32, 71, 72] (Table 6). The trials reported on the following outcomes: mortality, failure of clinical improvement (measured using a 7-point scale or hospital discharge), need for mechanical ventilation, and adverse events leading to treatment discontinuation.
Benefits
Among persons exposed to COVID-19, prophylactic treatment with lopinavir/ritonavir failed to show or exclude a beneficial effect on symptomatic SARS-CoV-2 infection, either independent of baseline PCR/serology or among those with a negative PCR and serology at baseline (HR: 0.60; 95% CI: 0.29, 1.26; moderate CoE and HR: 0.59; 95% CI: 0.17, 2.02; moderate CoE, respectively).
Among ambulatory patients with mild-to-moderate COVID-19, lopinavir/ritonavir failed to show or excluded a beneficial effect on COVID-19-related hospitalizations or deaths (HR: 1.16; 95% CI: 0.53, 2.56; moderate CoE and HR: 1.86; 95% CI 0.17 to 20.4; low certainty evidence, respectively).
Among hospitalized patients with COVID-19, treatment with lopinavir/ritonavir failed to show or exclude a beneficial effect on mortality or need for invasive mechanical ventilation (RR: 1.00; 95% CI: 0.89, 1.13; moderate CoE and RR: 1.12; 95% CI: 0.93, 1.34; low CoE). Similarly, lopinavir/ritonavir may reduce failure of clinical improvement at 14 days, but it is uncertain (RR: 0.78; 95% CI: 0.63, 0.97; very low CoE).
Harms
Prophylactic treatment of persons exposed to SARS-CoV-2 with lopinavir/ritonavir compared to placebo increases the risk of adverse events (RR: 2.74; 95% CI: 2.05, 3.66; moderate CoE). The most common adverse events were nausea/vomiting, diarrhea, abdominal pain, lack of appetite, itching and bloating.
Treatment of COVID-19 in ambulatory persons with lopinavir/ritonavir rather than placebo may increase the risk of serious adverse events (RR: 1.58; 95% CI: 0.79, 3.16; moderate CoE). RECOVERY reported 1/1588 serious adverse event due to treatment with lopinavir/ritonavir [72]; however, nearly 14% of lopinavir/ritonavir recipients in Cao 2020 were unable to complete the full 14-day course of administration. This was due primarily to gastrointestinal adverse events, including anorexia, nausea, abdominal discomfort, or diarrhea, as well as two serious adverse events, both acute gastritis. Two recipients had self-limited skin eruptions. Such side effects, including the risks of hepatic injury, pancreatitis, more severe cutaneous eruptions, and QT prolongation, and the potential for multiple drug interactions due to CYP3A inhibition, are well documented with this drug combination. The side effect profile observed in these trials raise concerns about the use of higher or more prolonged lopinavir/ritonavir dose regimens in efforts to improve outcomes.
Other considerations
The panel determined the certainty of evidence to be moderate due to concerns with imprecision for most critical outcomes across indications. The guideline panel made a strong recommendation against treatment with the combination of lopinavir/ritonavir for post-exposure prophylaxis, and ambulatory as well as hospitalized patients with COVID-19.
Conclusions and research needs for this recommendation
The guideline panel recommends against treatment with lopinavir/ritonavir across patient groups at risk for or with COVID-19.

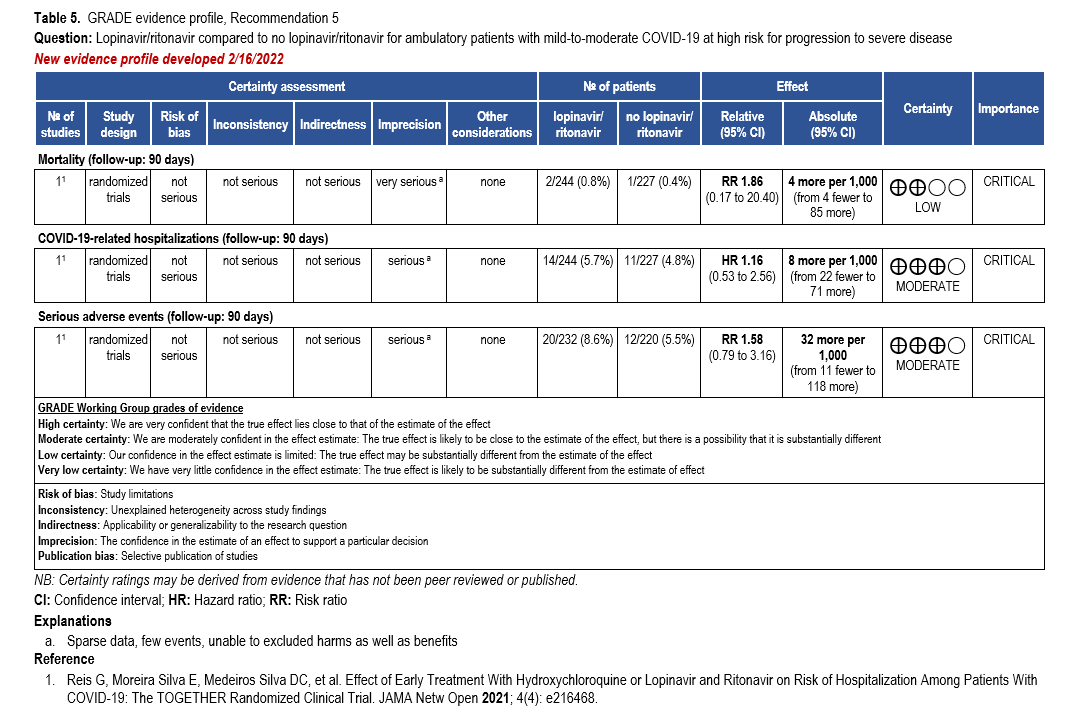
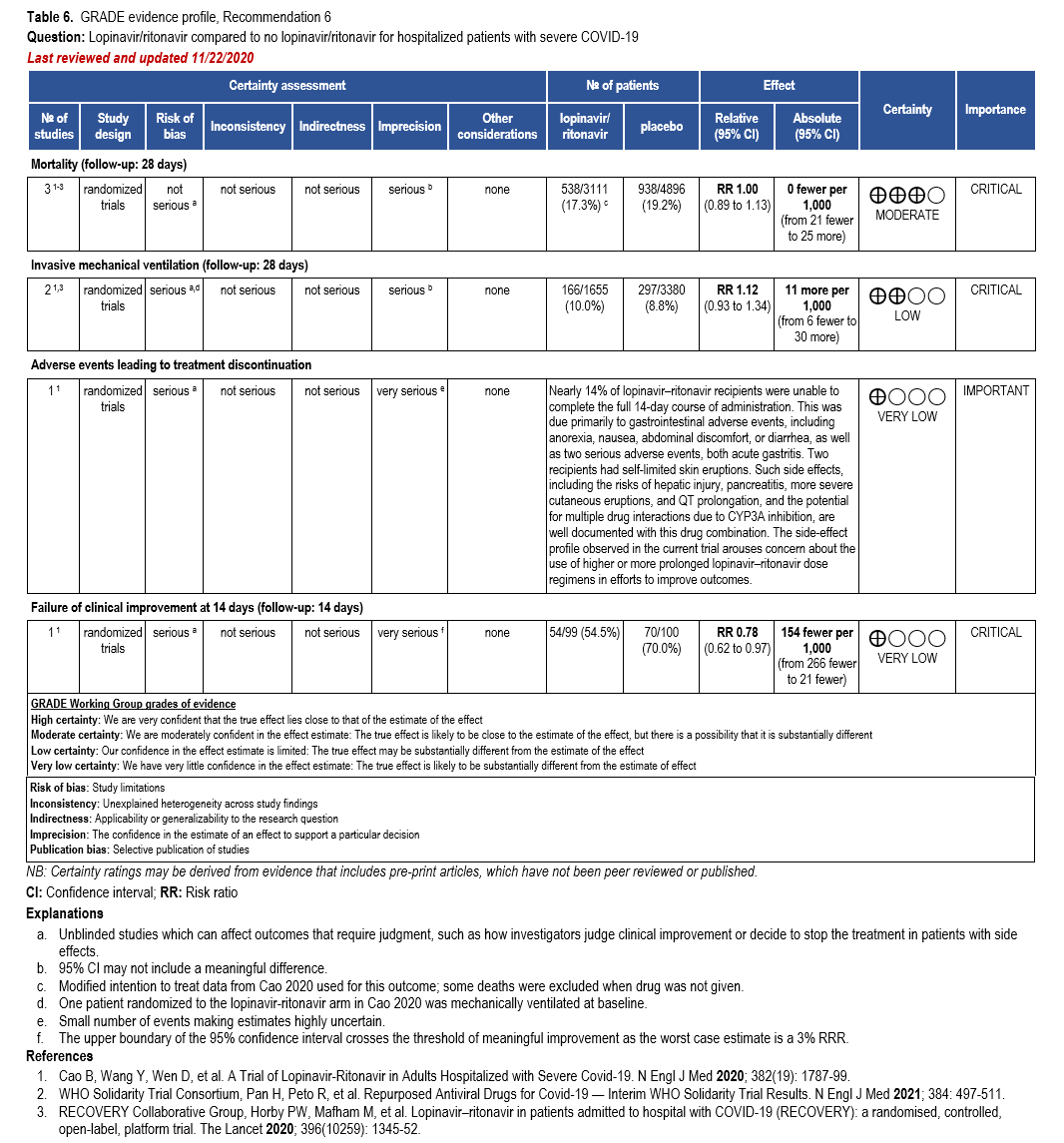
Supplementary Information
Study characteristics:
Forest plots:
- Figure s4a. Outcome of mortality at 28 days for lopinavir/ritonavir vs. no lopinavir/ritonavir
- Figure s4b. Outcome of invasive mechanical ventilation for lopinavir/ritonavir vs. no lopinavir/ritonavir
Risk of bias:
Recommendations 7-9: Glucocorticoids
Section last reviewed and updated 9/25/2020
Last literature search conducted 9/4/2020
Recommendation 7: Among hospitalized critically ill patients* with COVID-19, the IDSA guideline panel recommends dexamethasone rather than no dexamethasone. (Strong recommendation, Moderate certainty of evidence)
- Remark: If dexamethasone is unavailable, equivalent total daily doses of alternative glucocorticoids may be used. Dexamethasone 6 mg IV or PO for 10 days (or until discharge) or equivalent glucocorticoid dose may be substituted if dexamethasone unavailable. Equivalent total daily doses of alternative glucocorticoids to dexamethasone 6 mg daily are methylprednisolone 32 mg and prednisone 40 mg.
Recommendation 8: Among hospitalized patients with severe**, but non-critical, COVID-19, the IDSA guideline panel suggests dexamethasone rather than no dexamethasone. (Conditional recommendation†, Moderate certainty of evidence)
- Remark: Dexamethasone 6 mg IV or PO for 10 days (or until discharge) or equivalent glucocorticoid dose may be substituted if dexamethasone unavailable. Equivalent total daily doses of alternative glucocorticoids to dexamethasone 6 mg daily are methylprednisolone 32 mg and prednisone 40 mg.
Recommendation 9: Among hospitalized patients with mild-to-moderate*** COVID-19 without hypoxemia requiring supplemental oxygen, the IDSA guideline panel suggests against the use of glucocorticoids. (Conditional recommendation††, Low certainty of evidence)
Severity definitions:
- *Critical illness is defined as patients on mechanical ventilation and ECMO. Critical illness includes end organ dysfunction as is seen in sepsis/septic shock. In COVID-19, the most commonly reported form of end organ dysfunction is ARDS
- **Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
- ***Mild-to-moderate illness is defined as patient with a SpO2 >94% not requiring supplemental oxygen.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
The last literature search was conducted on September 4, 2020, and we identified eight RCTs and seven comparative non-randomized studies.
Why are corticosteroids considered for treatment?
In the early days of the SARS-CoV-2 pandemic, based on experience in both SARS and MERS, recommendations [73] cautioned against the use of systemic corticosteroids due to risk of worsening clinical status, delayed viral clearance, and adverse events [74-76]. Given the hyper-inflammatory state in COVID-19, immunomodulatory approaches, including steroids, continue to be evaluated to address both ARDS and systemic inflammation. ARDS stemming from dysregulated systemic inflammation may translate into prolonged ventilatory requirements and in-hospital mortality. In non-viral ARDS settings, there is increasing support for the role of steroids in the management of ARD [77]. A recent multicenter RCT in patients with moderate to severe ARDS demonstrated a reduced number of ventilatory days and reduction in mortality with use of a 10-day regimen of dexamethasone [78].
Summary of the evidence
Critical illness
Our search identified one systematic review that analyzed eight RCTs reporting on treatment with glucocorticoids among 1,844 critically ill patients with COVID-19 [79]. Three RCTs reported on patients treated with low- and high-dose dexamethasone [78, 80, 81]; three RCTs reported on patients treated with low-dose hydrocortisone [82-84]; and two RCTs reported on patients treated with high-dose methylprednisolone [79, 85]. The definition of critically ill varied across trials; however, the majority of patients had ARDS.
Severe and mild-to-moderate illness
Our search identified one RCT, one “partially” randomized trial, one prospective cohort, and five retrospective cohort studies [80, 86-92]. The RCT provided the best available evidence on treatment with corticosteroids for persons with COVID-19 [80] (Tables 7-9). Corral-Gudino et al. reported on a study that randomized patients to receive methylprednisolone or standard of care; however, patients expressing a preference for methylprednisolone were assigned to the same treatment arm [86]. Corral-Gudino et al. did not report the disaggregated results from the randomized trial; therefore, succumbing to the same potential for bias as reported subsequently for the non-randomized studies. The non-randomized studies had significant limitations with controlling for multiple co-interventions and disease severity at baseline [87-92]. All non-randomized studies had concerns with risk of bias due to lack of adjustment for critical confounders or potential for residual confounding. Timing of receipt, dose and duration of corticosteroids varied across studies.
The RECOVERY trial is a randomized trial among hospitalized patients in the United Kingdom [80]. In that study, 2,104 participants were randomized to receive dexamethasone (6 mg daily for up to 10 days) and 4,321 were randomized to usual care. The RECOVERY trial reported on the outcomes of mortality and hospital discharge. Participants and study staff were not blinded to the treatment arms.
Benefits
Critical illness
Among hospitalized, critically ill patients, the odds of mortality at 28 days was 34% less among patients treated with glucocorticoids than among patients not treated with glucocorticoids (OR: 0.66; 95% CI: 0.54; 0.82; high CoE). In addition, at 28 days, patients receiving dexamethasone were more likely to be discharged from the hospital (RR: 1.11; 95% CI: 1.04, 1.19; moderate CoE).
Severe illness
Among hospitalized patients, 28-day mortality was 17% lower in the group that received dexamethasone than in the group that did not receive dexamethasone (RR 0.83; 0.74-0.92; moderate CoE). In addition, at 28 days, patients receiving dexamethasone were more likely to be discharged from the hospital (RR: 1.11; 95% CI: 1.04, 1.19; moderate CoE).
Mild-to-moderate illness
In a sub-group analyses of patients without hypoxia not receiving supplemental oxygen, there was no evidence for benefit and a trend toward harm with dexamethasone in participants who were not on supplemental oxygen (RR 1.22; 0.86, 1.75; low CoE).
Harms
A systematic review of six studies did not report a difference in the events of serious adverse events experienced by patients randomized to receive treatment with glucocorticoids or no treatment with glucocorticoids (64/354 among those receiving glucocorticoids versus 80/342 among those not receiving glucocorticoids).
Patients receiving a short course of steroids may experience hyperglycemia, neurological side effects (e.g., agitation/confusion), adrenal suppression, and risk of bacterial and fungal infection [87, 93, 94].
Other considerations
Critical illness
The panel agreed that the overall certainty of the evidence for treatment with glucocorticoids for patients with critical COVID-19 was moderate due to concerns with indirectness and imprecision.
Severe illness
The panel agreed the overall certainty of evidence for treatment with glucocorticoids for patients with severe COVID-19 as moderate due to concerns with indirectness since the evidence was from dexamethasone.
Mild-to-moderate illness
The panel agreed that the overall certainty of evidence for patients without hypoxemia requiring supplemental oxygen as low due to concerns with risk of bias (post hoc analysis) and imprecision.
The panel agreed the overall certainty of evidence for treatment with glucocorticoids for patients with severe COVID-19 as moderate due to concerns with indirectness since the evidence was from dexamethasone. The panel agreed that the overall certainty of evidence for patients without hypoxemia requiring supplemental oxygen as low due to concerns with risk of bias (post hoc analysis) and imprecision.
Conclusions and research needs for these recommendations
The guideline panel recommends dexamethasone for patients with critical COVID-19. The guideline panel suggests dexamethasone for patients with severe COVID-19. If dexamethasone is not available, then alternative glucocorticoids may be used (see details above). The guideline panel suggests against glucocorticoids for patients with COVID-19 without hypoxemia requiring supplemental oxygen.
Additional research is needed to inform the generalizability of treatment with different glucocorticoids for patients with COVID-19 (Supplementary Table s2).
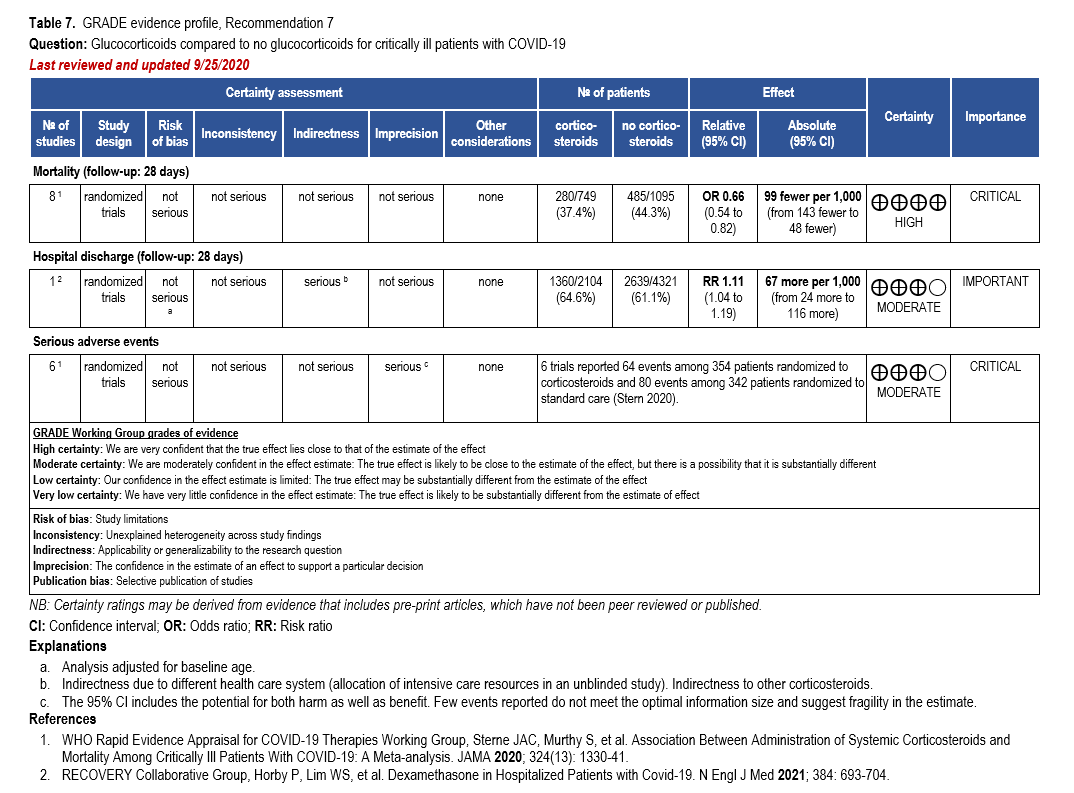
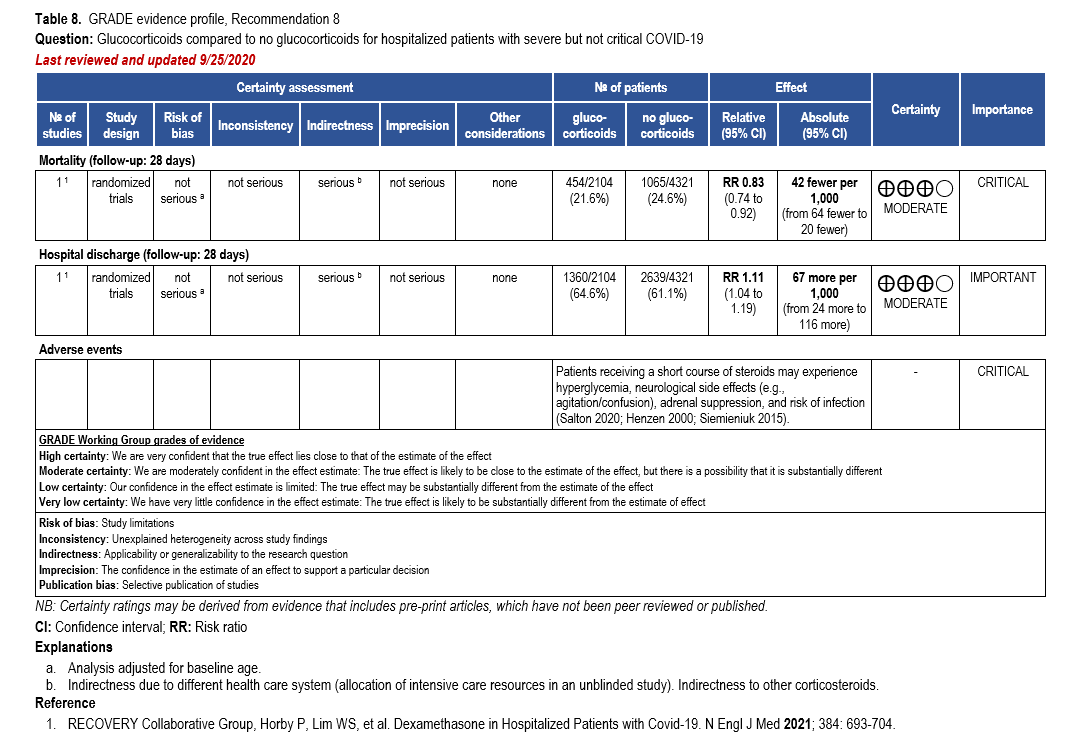
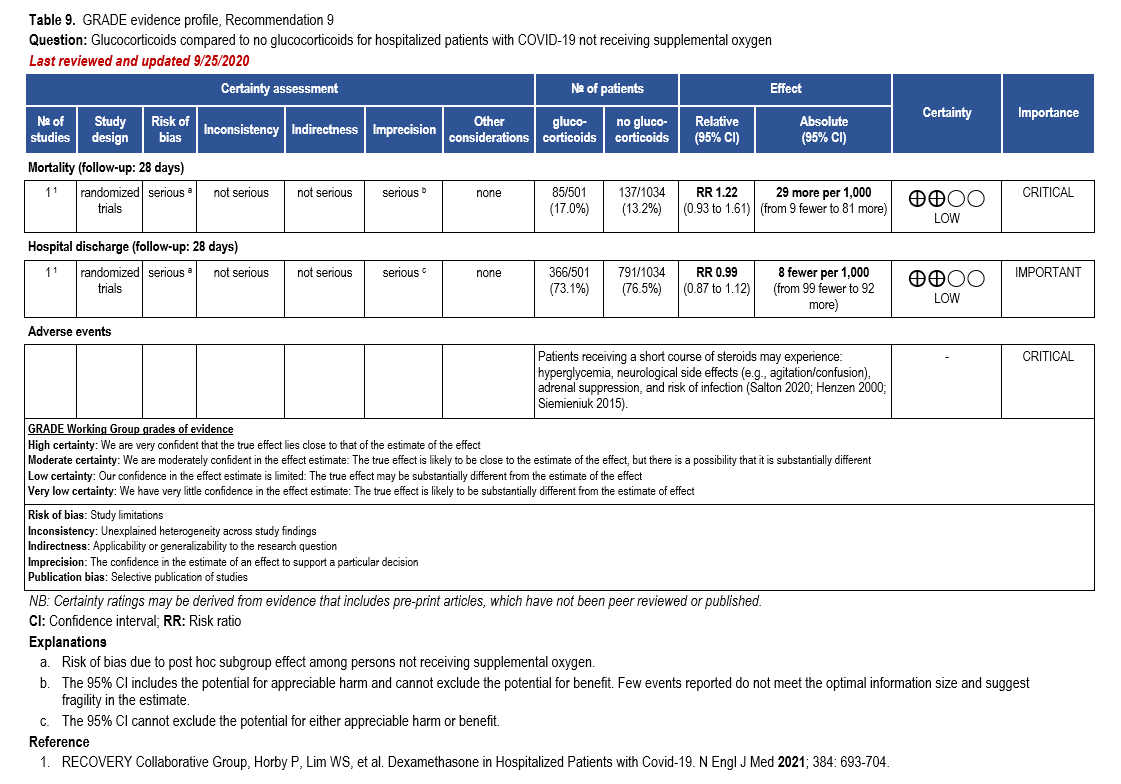
Supplementary Information
Study characteristics:
Risk of bias:
Recommendation 10: Inhaled corticosteroids
Section last reviewed and updated 10/10/2022
Last literature search conducted 8/31/2022
Recommendation 10: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA guideline panel suggests against inhaled corticosteroids. (Conditional recommendation††, Moderate certainty of evidence)
- Remark: Patients who are on inhaled corticosteroids for other indications may continue them.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why are inhaled corticosteroids considered for treatment?
Systemic corticosteroids have become a mainstay of therapy for the management of systemic inflammation seen in patients with severe COVID-19 infection as a result of the mortality reduction demonstrated in the RECOVERY trial [95]. In addition to their anti-inflammatory properties, some corticosteroids have been shown to inhibit viral replication of coronaviruses including MERS-CoV. Specifically, ciclesonide has demonstrated the ability to block SARS-CoV-2 viral replication in vitro, where fluticasone and dexamethasone did not [96]. Therefore, ciclesonide, and potentially other corticosteroids, may offer both anti-inflammatory and antiviral activity for the management of SARS-CoV-2. The antiviral mechanism may be related to the action of corticosteroids on both angiotensin converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2), which mediate SARS-CoV-2 viral attachment and entry into host cells. Preliminary data from a clinical cohort of patients taking inhaled corticosteroids suggest a lower expression of ACE2 and TMPRSS2 compared to those not taking inhaled corticosteroids and may suggest decreased susceptibility to SARS-CoV-2 in those taking inhaled corticosteroids [97].
Summary of the evidence
Eight randomized controlled trials (RCTs) reported on the use of inhaled corticosteroids budesonide, ciclesonide, or fluticasone compared to placebo or no treatment with inhaled corticosteroids for ambulatory or hospitalized patients with mild-to-moderate COVID-19 [98-105]. These trials reported on the outcomes of mortality, COVID-19-related hospitalization, and serious adverse events.
Benefits
Among patients with mild-to-moderate COVID-19, inhaled corticosteroids failed to show or exclude a beneficial effect on mortality or hospitalization (risk ratio [RR]: 0.58; 95% confidence interval [CI]: 0.24, 1.44; absolute risk reduction: 3 fewer per 1,000 [from 5 fewer to 3 more], moderate certainty of evidence [CoE] and RR: 0.81; 95% CI: 0.52, 1.27, low CoE).
Harms
Serious adverse events may be more frequent among patients with mild-to-moderate disease receiving treatment with inhaled corticosteroids rather than no inhaled corticosteroids; however, this may not be meaningfully different from those not receiving inhaled corticosteroids (RR: 1.14; 95% CI: 0.32, 3.99; moderate CoE).
Other considerations
The panel determined the certainty of evidence of treatment of inhaled corticosteroids for patients with mild-to-moderate COVID-19 to be moderate due to concerns with imprecision, as effects failed to show or exclude a beneficial effect for mortality or COVID-19-related hospitalization. The guideline panel made a conditional recommendation against inhaled corticosteroids outside of the context of a clinical trial.
Conclusions and research needs for this recommendation
The guideline panel suggests against inhaled corticosteroids for the treatment of patients with mild-to-moderate COVID-19. More information is needed about the interaction of inhaled corticosteroids with a 5-day course of ritonavir as part of nirmatrelvir/ritonavir treatment. When potent CYP 3A4 pharmacokinetic boosters like ritonavir or cobicistat are utilized for durations greater than 5 days in patients with HIV or hepatitis C, most inhaled corticosteroids are not recommended for coadministration due to the risk of Cushing’s syndrome and adrenal suppression [106]. This may be a consideration when prescribing inhaled steroids if concomitantly used with nirmatrelvir/ritonavir.
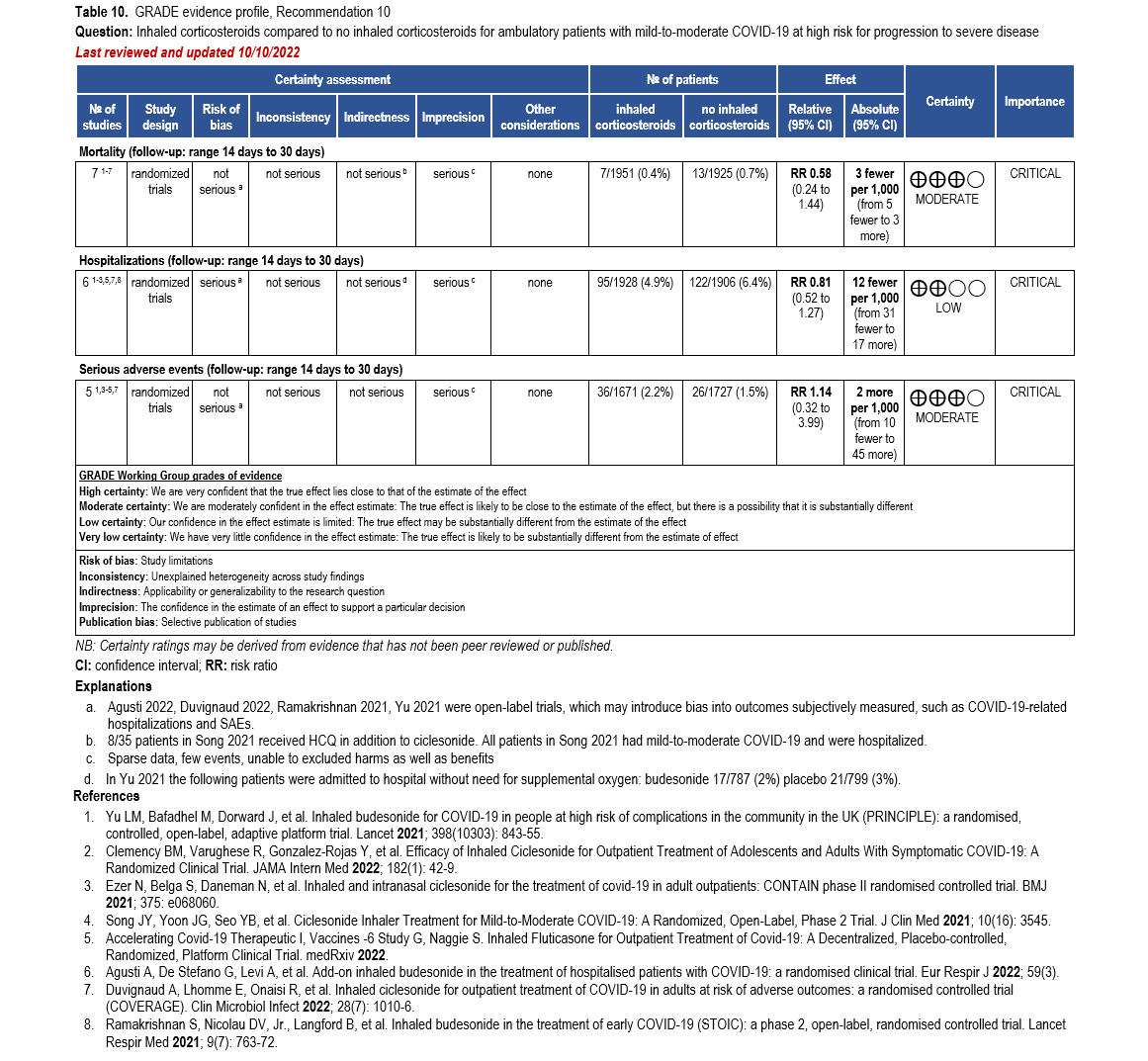
SUPPLEMENTARY MATERIALS
Study characteristics
Forest Plots
- Figure s5a. Mortality for inhaled corticosteroids compared to no inhaled corticosteroids in patients with mild-to-moderate COVID-19
- Figure s5b. Hospitalization for inhaled corticosteroids compared to no inhaled corticosteroids in patients with mild-to-moderate COVID-19
- Figure s5c. Serious adverse events for inhaled corticosteroids compared to no inhaled corticosteroids in patients with mild-to-moderate COVID-19
Risk of bias
Recommendations 11-12: IL-6 inhibitors (tocilizumab and sarilumab)
Section last reviewed and updated on 9/14/2021
Last literature search conducted 8/31/2021
Recommendation 11: Among hospitalized adults with progressive severe* or critical** COVID-19 who have elevated markers of systemic inflammation, the IDSA guideline panel suggests tocilizumab in addition to standard of care (i.e., steroids) rather than standard of care alone. (Conditional recommendation†, Low certainty of evidence)
Remarks:
- Patients, particularly those who respond to steroids alone, who put a high value on avoiding possible adverse events of tocilizumab and a low value on the uncertain mortality reduction, would reasonably decline tocilizumab.
- In the largest trial on the treatment of tocilizumab, criterion for systemic inflammation was defined as CRP ≥75 mg/L.
Recommendation 12: When tocilizumab is not available for patients who would otherwise qualify for tocilizumab, the IDSA guideline panel suggests sarilumab in addition to standard of care (i.e., steroids) rather than standard of care alone. (Conditional recommendation†, Very low certainty of evidence)
- Remark: Patients, particularly those who respond to steroids alone, who put a high value on avoiding possible adverse events of sarilumab and a low value on the uncertain mortality reduction, would reasonably decline sarilumab.
Severity definitions:
- *Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
- **Critical illness is defined as patients on mechanical ventilation and ECMO. Critical illness includes end organ dysfunction as is seen in sepsis/septic shock. In COVID-19, the most commonly reported form of end organ dysfunction is ARDS.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why are are interleukin-6 (IL-6) receptor antagonists considered for treatment?
Some patients with COVID-19 develop a hyperinflammatory syndrome that is characterized by elevations in proinflammatory cytokines and multiorgan dysfunction also known as the immunopathology of SARS-CoV-2 infection. The significance of these findings is unclear, however early descriptions found that those with elevated IL-6 levels and evidence of hyperinflammation had increased rates of more severe disease [107, 108]. Tocilizumab, a monoclonal anti-IL-6-receptor blocking antibody, has been proposed as a therapeutic agent to mitigate hyperinflammation associated with COVID-19. Tocilizumab is FDA-approved for various rheumatologic conditions as well as cytokine release syndrome associated with CAR-T cell therapy.
Sarilumab, another IL-6 receptor antagonist, is currently FDA-approved for rheumatoid arthritis (RA).
Summary of Evidence
Tocilizumab
Our search identified eight RCTs (including pre-prints) that reported on patients with severe COVID-19 randomized to treatment with tocilizumab (8 mg/kg) or placebo/usual care [109-116]. Gordon 2020, Horby 2021, Rosas 2020, and Veiga 2021 allowed for patients to be on mechanical ventilation at randomization, whereas the other trials included patients with a lower disease severity (e.g., allowed supplemental oxygen but excluded those on higher levels of oxygen support) or included patients with severe COVID with an inflammatory phenotype.
One trial, RECOVERY, contributed the majority of the weight in the analysis [111]. RECOVERY trial participants must have demonstrated clinical evidence of progressive COVID-19, which was defined as <92% oxygen saturation on room air or receiving oxygen and C-reactive protein (CRP) ≥75 mg/L. Use of steroids was balanced across both the participants receiving tocilizumab or not receiving tocilizumab. Following recommendations for treatment with glucocorticoids, 82% of participants in both arms received dexamethasone. While RECOVERY did not blind participants or healthcare personnel to the randomized treatment arm, this likely would not introduce bias in the objective measurement of the outcome of mortality; however, it was considered as a risk of bias for more subjectively measured outcomes, clinical deterioration, along with the total body of evidence contributing to those outcomes (Table 11). There are limited safety data in the preliminary report.
Both RECOVERY and REMAP CAP (the two tocilizumab trials that reported a benefit) initiated treatment early (randomization at median of two days of hospitalization in RECOVERY; <24 hours in the ICU for REMAP-CAP), suggesting tocilizumab may be more beneficial early in people with rapidly progressive disease.
Sarilumab
We identified three RCTs that reported on patients with severe or critical COVID-19 randomized to treatment with sarilumab or placebo/usual care [109, 117, 118]. In addition, a pre-print network meta-analysis of 18 RCTs was identified that reported network estimates for sarilumab plus corticosteroids compared with usual care alone [119].
Benefits
Tocilizumab
Among hospitalized patients, tocilizumab showed a trend toward reduced mortality at 28 days compared to no tocilizumab treatment (RR: 0.91; 95% CI: 0.79, 1.04; moderate CoE). Tocilizumab demonstrated a lower relative risk of clinical deterioration, defined as death, need for mechanical ventilation, ECMO, or ICU admission, compared to placebo/usual care, RR: 0.83 (95% CI: 0.77, 0.89; moderate CoE). Four studies were not blinded, while in the remaining three trials healthcare personnel and outcome assessors were blinded. The panel noted that tocilizumab causes a decline in CRP levels, which if obtained would reveal the treatment arm designations of the patients, therefore introducing bias for the more subjectively measured outcomes of clinical deterioration and serious adverse events.
Sarilumab
Among hospitalized patients, sarilumab showed a trend toward reduced mortality at 28 days compared to usual care (network estimate OR: 0.80; 95% CI: 0.61, 1.04; low certainty of evidence). Sarilumab may reduce clinical deterioration, defined as progression to intubation, ECMO or death compared to usual care (RR: 0.67; 95% CI: 0.42, 1.05; very low CoE).
Harms
Serious adverse events among patients receiving tocilizumab or sarilumab did not differ from those receiving usual care (RR: 0.89; 95% CI: 0.74, 1.07; low CoE and RR: 1.03; 95% CI: 0.89, 1.18; low CoE, respectively). An additional trial attributed treatment with tocilizumab to three serious adverse events; however, did not report events among patients not receiving tocilizumab [111]. Previously, tocilizumab has been associated with gastrointestinal perforations in non-COVID-19 settings, and case reports of bowel perforations have recently emerged with the use of tocilizumab for COVID-19 [120-123]. Increased infection risks have been noted in uncontrolled studies, and it is possible that this risk may be compounded by the combination of glucocorticoids and tocilizumab. [124, 125].
Other considerations
While the overall certainty of evidence for the trend toward a reduction in mortality was moderate, the panel believes that differences in mortality rates across the trials may be the result of the differences in baseline severity of study participants and timing of tocilizumab receipt in the disease course. In REMAP-CAP, tocilizumab was administered within 24 hours of participants’ initiating organ support in an intensive care unit, raising the possibility that this may be the optimal time to administer the drug. In RECOVERY, tocilizumab was administered to participants with oxygen saturation <92% on room air or receiving oxygen therapy, and CRP ≥75 mg/L. Given the reduction in clinical deterioration and trend toward mortality reduction, the guideline panel made a conditional recommendation for treatment of adults with tocilizumab.
The use of tocilizumab, as with other therapeutic agents that can suppress the immune system, presents additional considerations and potential concerns when used in immunocompromised hosts. The panel did not conduct an analysis of available data to assess differences in efficacy and/or adverse effects of tocilizumab among oncology or other immunocompromised patients at this time.
The panel recognized the current shortage of tocilizumab and possible net benefit of treatment with sarilumab.
Conclusions and research needs for this recommendation
The guideline panel suggests tocilizumab for hospitalized adults with COVID-19. When tocilizumab is not available and baricitinib is either not appropriate or available, the guideline panel suggests sarilumab for persons who would otherwise qualify for tocilizumab; however, it is acknowledged that patients, particularly those responding to steroids alone or baricitinib, who put a high value on avoiding the possible adverse events of sarilumab and a low value on the uncertain mortality reduction would reasonably decline sarilumab.
Additional research is needed to understand the efficacy of tocilizumab when taken at different times during the course of disease. For example, there are no data to guide recommendations in patient <18 years of age at this time. In addition, future studies are needed to inform the generalizability of tocilizumab with different IL-6 receptor inhibitors for patients with COVID-19 (Supplementary Table s2). At the time of update, preliminary data from a trial of treatment with sarilumab has been shared as a pre-print [109]; however, number of patients who received sarilumab is limited (n=45) and the published manuscript was not available for analysis or inclusion to inform this recommendation. Other studies of sarilumab have not been made available.


Supplementary Information
Study characteristics:
Forest plots:
- Figure s6a. Outcome of mortality for tocilizumab vs. no tocilizumab
- Figure s6b. Outcome of mortality for tocilizumab vs. no tocilizumab (sensitivity analysis for patients on mechanical ventilation for <24 hours)
- Figure s6c. Outcome of clinical deterioration for tocilizumab vs. no tocilizumab
- Figure s6d. Outcome of severe adverse events for tocilizumab vs. no tocilizumab
Risk of bias:
Recommendations 13-15: Convalescent plasma
Section last reviewed and updated on 2/22/2023
Last literature search conducted 1/31/2023
Recommendation 13(UPDATED 2/22/2023): Among immunocompetent patients hospitalized with COVID-19, the IDSA guideline panel recommends against COVID-19 convalescent plasma. (Strong recommendation, Moderate certainty of evidence).
Recommendation 14(NEW 2/22/2023): Among immunocompromised patients hospitalized with COVID-19, the IDSA guideline panel suggests against the routine use of COVID-19 convalescent plasma. (Conditional recommendation, very low certainty of evidence)
Remarks:
- Patients, particularly those who do not qualify for other treatments, who place a higher value on the uncertain mortality reduction and a lower value on the potential adverse effects of convalescent plasma would reasonably select convalescent plasma.
Recommendation 15(UPDATED 2/22/2023): Among ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options*, the IDSA guideline panel suggests FDA-qualified high-titer COVID-19 convalescent plasma within 8 days of symptom onset rather than no high-titer COVID-19 convalescent plasma. (Conditional recommendation†, Low certainty of evidence)
*Other options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir and three-day treatment with remdesivir Patient-specific factors (e.g., symptom duration, renal insufficiency or other contraindications, drug interactions) as well as logistical challenges, infusion capacity, and product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Remarks:
- In the United States, FDA emergency use authorization (EUA) only authorizes use in patients with immunosuppressive disease or receiving immunosuppressive treatment.
- Patients, particularly those who are not immunocompromised, who place a low value on the uncertain benefits (reduction in the need for mechanical ventilation, hospitalization, and death) and a high value on avoiding possible adverse events associated with convalescent plasma would reasonably decline convalescent plasma.
Why is convalescent plasma considered for treatment?
Convalescent plasma has been used as passive immunotherapy for prevention and treatment of infections for over 100 years [126, 127]. The predominant proposed protective mechanism is thought to be pathogen neutralization, although antibody-dependent cellular cytotoxicity and enhanced phagocytosis may also play a role. With the advent of effective antimicrobial therapy (i.e., “the antibiotic era”), convalescent plasma fell out of favor. In recent years, interest in this approach has revived as a means of addressing viral epidemics such as Ebola, SARS-CoV-1, and MERS. Studies of convalescent plasma derived from people who had recovered from those specific infections showed encouraging results but were typically small, non-randomized, and largely descriptive [128-130].
In the current pandemic, convalescent plasma obtained from individuals who have recovered from COVID-19 has been used in over 100,000 patients with moderate to severe infection as part of an expanded access program (EAP) [131, 132]. In an analysis of the convalescent plasma EAP, higher levels of antibodies were associated with significant improvements in mortality compared to receipt of convalescent plasma with lower concentrations of neutralizing antibodies [131]. However, there was no placebo group in the study. Subgroup analysis from one open-label randomized controlled trial [RCT] reporting on plasma with anti-receptor-binding domain ELISA values corresponding to a high antibody titer cutoff showed a non-significant relative risk reduction in mortality of 5% (Risk ratio [RR]: 0.95; 95% confidence interval [CI]: 0.73, 1.25) [133]. An additional subgroup analysis suggested unselected convalescent plasma (i.e., not limited to high-titer antibodies) may increase the relative risk for mortality by 49% (RR: 1.42; 95% CI: 0.92, 1.69).
An analysis of the convalescent plasma EAP suggested greatest benefit when convalescent plasma is given within three days from diagnosis [131]. In August 2020, the FDA issued an EUA for investigational convalescent plasma for the treatment of COVID-19 in hospitalized patients [134]. In early February 2021, the FDA issued a revision to the EUA to limit authorization to the use of high-titer COVID-19 convalescent plasma for treatment of hospitalized patients early in the disease course [135].
Summary of the evidence
Our search identified and was informed by evidence from 23 RCTs and a large (n=20,000), single-arm registry study [126-130, 136-147], as they provided the best available evidence for the outcomes of mortality, need for mechanical ventilation, serious adverse events, and adverse events. Eighteen of those RCTs reported on convalescent plasma for patients hospitalized with COVID-19 (Table 13) [126-129, 136-141], two RCTs (Denkinger & Hueso) reported on receipt of convalescent plasma by immunocompromised patients hospitalized with COVID-19 (Table XX), and three RCTs [143-145] reported on receipt of convalescent plasma by ambulatory persons with mild COVID-19 disease (Table 15).
Eighteen trials randomized 17,232 patients hospitalized with COVID-19 to receive COVID-19 convalescent plasma [126-129, 136-141]. Several trials were open-label and/or had concerns with risk of bias due to lack of adjustment for critical confounders or potential for residual confounding (Supplementary Table s16a). Timing of receipt of COVID-19 convalescent plasma during the clinical course of the patients’ illness varied across studies (Supplementary Table s15). One trial reported on 160 persons who received high-titer convalescent plasma less than 72 hours after the onset of symp-toms of COVID-19 (mean age: 77.2 years; standard deviation: ±8.6 years) [130]. In addition, Joyner 2020 reported on safety outcomes of over 20,000 patients enrolled in the convalescent plasma EAP.
Benefits
Hospitalized patients
In hospitalized patients, , convalescent plasma appears to have trivial little or no effect on mortality based on the body of evidence from RCTs (RR: 0.98; 95% CI: 0.93, 1.03; moderate certainty of evidence [CoE]). Recipients of COVID-19 convalescent plasma may have a greater need for mechanical ventilation (RR: 1.10; 95% CI: 0.94, 1.29; low CoE); however, the evidence is uncertain because of concerns with risk of bias and imprecision.
In hospitalized immunocompromised patients, convalescent plasma failed to show or to exclude a beneficial effect on mortality based on the body of evidence from two RCTs (RR: 0.65; 95% CI: 0.37, 1.13; very low CoE).
Ambulatory persons
Receipt of COVID-19 convalescent plasma was associated with a reduction in hospitalization (RR: 0.74; 95% CI: 0.56, 0.98; moderate CoE) and a trend toward a reduction in COVID-19 related hospitalizations or medically attended visits (emergency room or urgent care; RR 0.79; 95% CI: 0.63 to 1.00; moderate CoE); however, the evidence remains uncertain due to few reported events. Similarly, evidence showed a possible reduction of progression to severe respiratory disease (RR: 0.52; 95% CI: 0.29, 0.94; low CoE); however, the evidence remains uncertain, as oxygenation and respiration rates are surrogate measures of need for ventilation, morbidity, and death, and because of the fragility of the estimate due to the small number of events reported. Convalescent plasma failed to show or exclude a beneficial effect on all-cause mortality based on the body of evidence from two RCTs (RR: 0.53; 95% CI: 0.14, 1.98; low CoE); however, the evidence is uncertain due to concerns with fragility of the estimate due to the small number of events reported. Additional deaths beyond 15 days were reported in one RCT and included five deaths in the plasma group versus one in the placebo arm.
Harms
In the largest safety study (n=20,000), within four hours of completion of convalescent plasma transfusion, authors reported 146 serious adverse events (SAEs) classified as transfusion reactions (<1% of all transfusions) [142]. Of these, 63 deaths were reported (0.3%), with 13 judged as possibly or probably related to the transfusion. The non-mortality SAEs include 37 reports of transfusion-associated circulatory overload, 20 cases of transfusion-related acute lung injury, and 26 cases of severe allergic transfusion reactions.
Within seven days of transfusion, 1711 deaths were reported (mortality rate: 8.56%; 95% CI: 8.18, 8.95). In addition, 1136 SAEs were reported: 643 cardiac events (569 judged as unrelated to the transfusion), 406 sustained hypotensive events requiring pressor support, and 87 thromboembolic or thrombotic events (55 judged as unrelated to the transfusion).
Eleven trials among patients hospitalized for COVID-19 suggest increased adverse events among patients receiving convalescent plasma (RR: 1.08; 95% CI: 0.94, 1.26; low CoE); however, the evidence was uncertain due to concerns with lack of blinding. In addition, included studies lacked a standard definition for what met the definition of an adverse event. In ambulatory patients, SAEs were higher in the convalescent plasma group due to serious transfusion reactions requiring treatment or admission (RR 5.95; 95% CI: 0.72, 49.29; low CoE), although the evidence is uncertain due to few events.
Immunocompromised recipients of COVID-19 convalescent plasma may experience a higher number of SAEs (RR: 1.20; 95% CI: 0.86, 1.68; low CoE); however, the evidence from two RCTs is uncertain because of concerns with risk of bias and imprecision.
Other considerations
Hospitalized patients
The panel agreed that the overall certainty of evidence is moderate due to some remaining imprecision as the 95% CI crossed the threshold of 1% for plausible mortality reduction. The guideline panel recognized that unselected use of convalescent plasma appeared to have trivial to no beneficial effect from the now existing large body of evidence. In the subgroup of immunocompromised patients, the panel agreed that very low certainty evidence failed to show or exclude a beneficial effect, mostly due to risk of bias and imprecision due to small number of events. In addition, studies were conducted in the pre-omicron, pre-vaccination era with a significantly higher baseline risk for a poor outcome, making the findings less applicable and more uncertain.
Ambulatory persons
The panel agreed that the overall certainty of evidence is low due to concerns with imprecision, which recognized the limited events and concerns with fragility. The guideline panel recognized the inability to exclude a meaningful beneficial or detrimental effect when convalescent plasma is given early in the course of COVID-19 disease.
Conclusions and research needs for this recommendation
Additional clinical trials may be needed to more definitively determine whether there is a benefit of treatment with COVID-19 convalescent plasma and at what dose (neutralizing antibody titers), especially for patients early in the disease course of COVID-19 (Supplementary Table s2).
Given the available evidence summarized above, the guideline panel suggests against COVID-19 convalescent plasma for persons hospitalized with COVID-19. Based on limited studies and mechanistic reasoning, COVID-19 convalescent plasma may be more effective if given at high titers early in course of hospitalization, in patients with undetectable or low levels of anti-SARS-CoV-2 antibodies, or in those with a humoral immune deficiency [148-153]. Current RCTs have not reported outcomes in such pre-specified subpopulations. Future studies in hospitalized patients should focus on patients with humoral immunodeficiencies early in the course of COVID-19. Future studies in hospitalized patients should also consider screening for SARS-CoV-2 neutralizing antibodies in all patients at entry into RCTs and assessing outcomes based on antibody levels.
The guideline panel suggests FDA-qualified high-titer COVID-19 convalescent plasma in the ambulatory setting for persons with mild-to-moderate COVID-19 at high risk for progression to severe disease, who have no other treatment options. In ambulatory patients, convalescent plasma may be more effective if the product used contains high titers of neutralizing antibodies and is used early in clinical presentation or in subpopulations of patients who do not have an adequate humoral immune response even at later stages of disease [148]. The existing evidence in this specific population of patients remains sparse. Future studies in ambulatory patients should continue to target these populations.

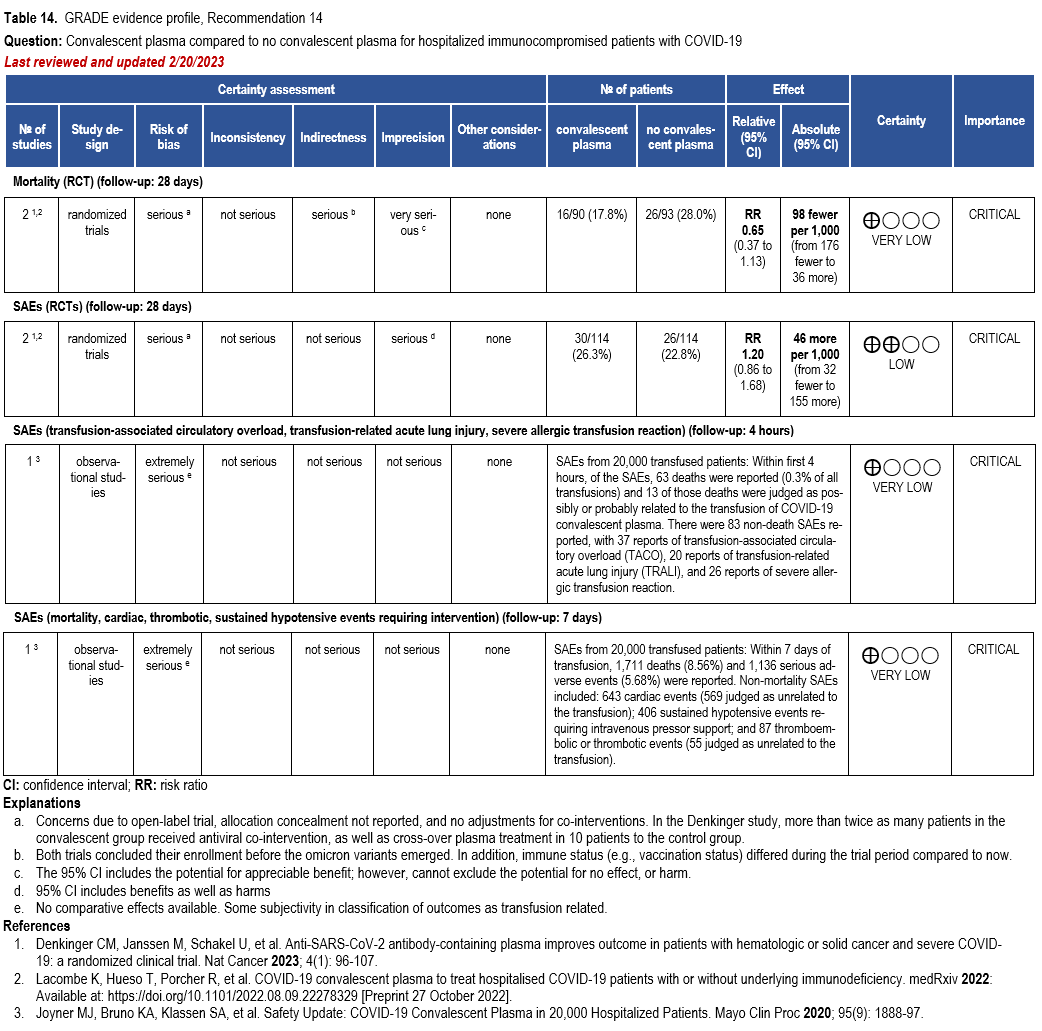
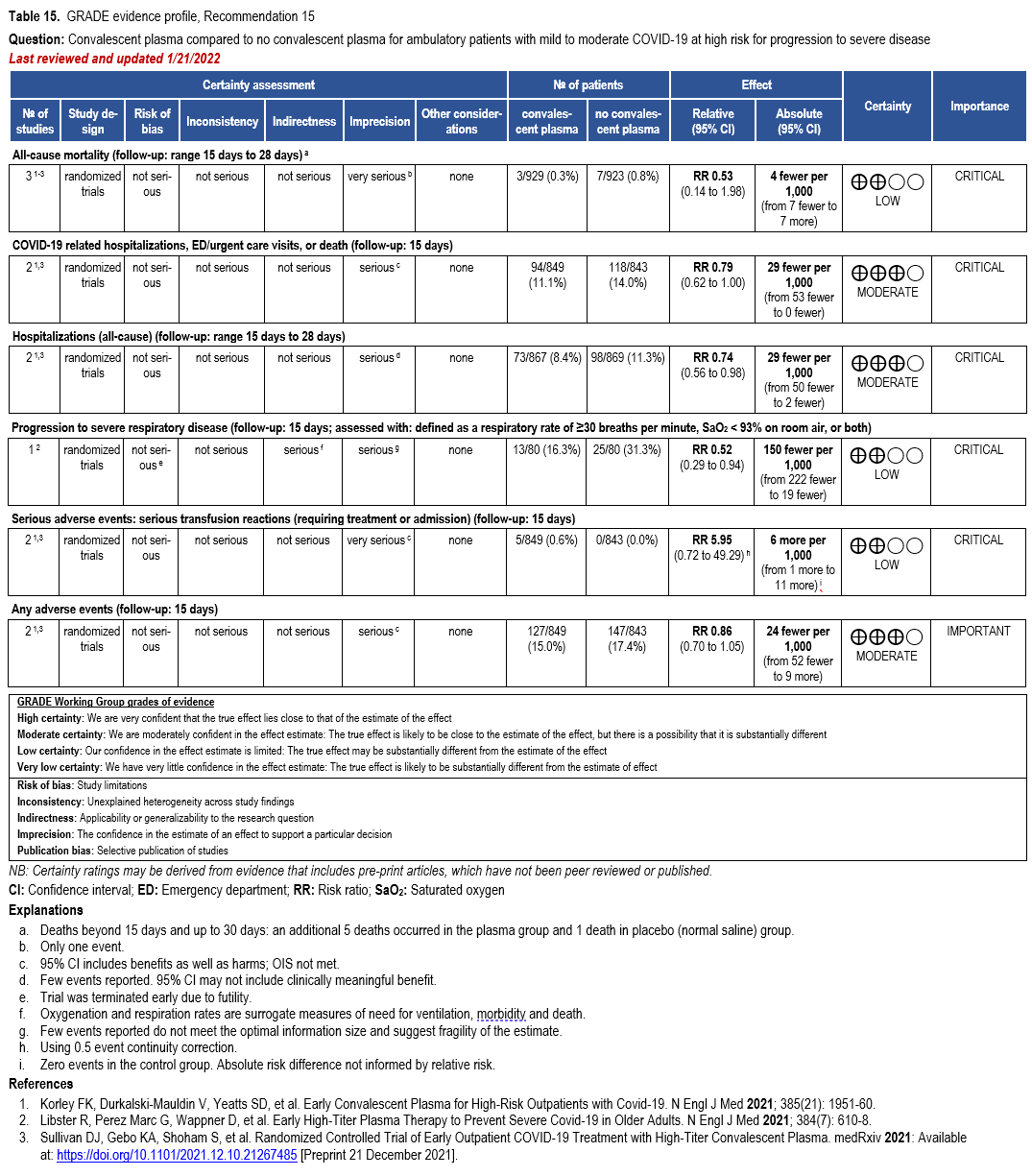
Supplementary Information
Study characteristics:
Forest plots:
- Figure s7a. Forest plot for the outcome of mortality for convalescent plasma vs. no convalescent plasma in hospitalized patients
- Figure s7b. Forest plot for the outcome of mechanical ventilation for convalescent plasma vs. no convalescent plasma in hospitalized patients
- Figure s7c. Forest plot for the outcome of adverse events (mild to severe) for convalescent plasma vs. no convalescent plasma in hospitalized patients
- Figure s7d. Forest plot for the outcome of mortality for convalescent plasma vs. no convalescent plasma in ambulatory patients
- Figure s7e. Forest plot for the outcome of COVID-19-related hospitalizations for convalescent plasma vs. no convalescent plasma in ambulatory patients
- Figure s7f. Forest plot for the outcome of all-cause hospitalizations for convalescent plasma vs. no convalescent plasma in ambulatory patients
- Figure s7h. Forest plot for the outcome of adverse events for convalescent plasma vs. no convalescent plasma in ambulatory patients
- Figure s7i. Forest plot for the outcome of mortality for convalescent plasma vs. no convalescent plasma in hospitalized immunocompromised patients
- Figure s7j. Forest plot for the outcome of SAEs for convalescent plasma vs. no convalescent plasma in hospitalized immunocompromised patients
Risk of bias:
- Table s16a. Risk of bias for randomized controlled studies (convalescent plasma vs. no convalescent plasma)
- Table s16b. Risk of bias for non-randomized studies (convalescent plasma vs. no convalescent plasma)
Recommendations 16-18: Remdesivir
Section last reviewed and updated 2/7/2022
Last literature search conducted 1/31/2022
Recommendation 16: Among patients (ambulatory or hospitalized) with mild-to-moderate COVID-19 at high risk for progression to severe disease, the IDSA guideline panel suggests remdesivir initiated within seven days of symptom onset rather than no remdesivir. (Conditional recommendation†, Low certainty of evidence)
Remarks:
- Dosing for remdesivir in mild-to-moderate COVID-19 is 200 mg on day one followed by 100 mg on days two and three. Pediatric dosing is 5 mg/kg on day 1 and 2.5 mg/kg on subsequent days.
- Options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, three-day treatment with remdesivir, molnupiravir, and neutralizing monoclonal antibodies. Patient-specific factors (e.g., patient age, symptom duration, renal function, drug interactions), product availability, and institutional capacity and infrastructure should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
Recommendation 17: In patients on supplemental oxygen but not on mechanical ventilation or ECMO, the IDSA panel suggests treatment with five days of remdesivir rather than 10 days of remdesivir. (Conditional recommendation†, Low certainty of evidence)
Recommendation 18a: In hospitalized patients with severe* COVID-19, the IDSA panel suggests remdesivir over no antiviral treatment. (Conditional recommendation†, Moderate certainty of evidence)
*Severe illness is defined as patients with SpO2 ≤94% on room air.
Recommendation 18b: In patients with COVID-19 on invasive ventilation and/or ECMO, the IDSA panel suggests against the routine initiation of remdesivir (Conditional recommendation††, Very low certainty of evidence)
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is remdesivir considered for treatment?
Remdesivir (GS-5734) is an antiviral drug with potent in vitro activity against a range of RNA viruses including MERS-CoV, SARS-CoV 1 & 2 [154-156]. Remdesivir acts by causing prem-ature termination of viral RNA transcription [156]. Its use improved disease outcomes and re-duced viral loads in SARS-CoV-1 infected mice [155]. In rhesus macaques, therapeutic treat-ment with remdesivir showed reduction in SARS-CoV-2 loads, pathologic changes, and progres-sion of clinical disease [157]. In this same animal model, remdesivir treatment initiated 12 hours post-inoculation reduced clinical signs, virus replication in the lungs, and decreased the presence and severity of lung lesions.
Summary of the evidence
Patients with mild-to-moderate disease who are at high risk for progression to severe COVID-19
One RCT compared treatment with three days of intravenous (IV) remdesivir (200 mg on day one followed by 100 mg on days two and three) initiated within 7 days of symptom onset or no remdesivir in unvaccinated patients [158]. The study enrolled patients at high risk for progression (e.g., obesity, diabetes mellitus, hypertension, immune compromise etc.) or age 60 years or older who were symptomatic seven days or less without prior treatment (e.g., mono-clonal antibodies), but were not expected to receive oxygen at time of enrollment (>94% on room air). The outcomes assessed were mortality, hospitalizations for any cause, and COVID-19-related medically as well as serious adverse events.
Hospitalized patients with SpO2 ≤94% on room air
Three RCTs comparing treatment with remdesivir (200 mg day one, 100 mg daily days 2-10) against no remdesivir treatment [32, 159, 160], and one RCT comparing five days of treatment (200 mg day one, 100 mg daily days 2-5) against 10 days (200 mg day one, 100 mg daily days 2-10) of treatment [161] served as the best available evidence among hospitalized persons with severe COVID-19 (Table 17-18). The outcomes assessed were mortality, time to clinical improvement, need for mechanical ventilation, serious adverse events, and adverse events leading to treatment discontinuation.
All trials used different definitions of severe disease for participants. ACTT-1 partici-pants were considered to have severe disease if they required mechanical ventilation, supple-mental oxygen, if SpO2 was 94% or lower while breathing ambient air, or if they had tachypnea (respiratory rate >24 breaths per minute) [159]. Within the SOLIDARITY trial (available only as a pre-print at this time), participants with severe disease were receiving mechanical ventilation [32]. In Wang 2020, severe participants had a SpO2 <94% while breathing room air or a ratio of arterial oxygen partial pressure to fractional inspired O2 of <300 mm Hg and radiologically con-firmed pneumonia.
Updated analyses include the final analysis from the ACTT-1 and the interim analysis of the SOLIDARITY trial [32, 159]. SOLIDARITY reported mortality among persons remaining in hospital up to the duration of the study; however, among patients discharged before the end of the study, mortality may not have been collected completely. The study by Wang et al (2020) was stopped early due to lack of recruitment into the trial due to decreased incidence in China.
Randomization performed in Goldman 2020 failed to establish prognostic balance be-tween baseline clinical status among the 397 patients randomized into the treatment arms, with patients in the 10-day arm more severely ill at study entry. Even with the adjusted analysis, residual confounding is possible. In addition, participants, healthcare workers, and outcome as-sessors were not blinded to the treatment arms.
Hospitalized patients on invasive ventilation and/or ECMO
Subgroups from SOLIDARITY and ACTT-1 reported on the outcomes of mortality, time to recovery and serious adverse events among patients on invasive ventilation or ECMO [32, 159] (Table 18b). The duration of ventilation at time of treatment with remdesivir was not reported in ACTT-1. This may introduce uncertainty when assessing outcomes of mortality or time to re-covery.
In ACTT-1 [159], randomization was stratified by study site and disease severity at en-rollment. Disease severity groups were mild-to-moderate COVID-19 (SpO2 >94%) and severe COVID-19 (SpO2 ≤94%). The severe COVID-19 stratum included patients who were hypoxemic with various degrees of severity including those requiring low flow oxygen by nasal cannula, those needing high-flow oxygen, non-invasive ventilation, invasive mechanical ventilation and ECMO. In addition to analyses on established strata, authors performed post hoc analyses for subgroups within the strata (e.g., receiving oxygen, receiving high-flow oxygen or noninvasive mechanical ventilation, or receiving mechanical ventilation or ECMO), which may introduce concerns with risk of bias and imprecision when making inferences on efficacy of remdesivir among these subgroups including mechanically ventilated patients.
Benefits
Patients with mild-to-moderate disease who are at high risk for progression to severe COVID-19
Treatment with remdesivir for three days in ambulatory patients reduced hospitalizations and COVID-19-related medically attended visits throughout day 28 (HR: 0.28; 95% CI: 0.1, 0.75, low CoE; and HR: 0.19; 95% CI: 0.07, 0.56, low CoE, respectively). No deaths were observed.
Hospitalized patients with SpO2 ≤94% on room air
The pooled analysis failed to show a mortality benefit at 28 days (RR: 0.92; 95% CI: 0.77, 1.10; low CoE) [32, 159, 160]. Patients receiving treatment with remdesivir trend toward greater clinical improvement at 28 days than patients not receiving remdesivir (RR: 1.13; 95% CI: 0.91, 1.41; low CoE) [160]. In addition, based on a post hoc analysis of patients with severe COVID-19, receiving treatment with remdesivir had a shorter median time to recovery (median 11 vs. 18 days; rate ratio: 1.31; 95% CI: 1.12, 1.52; low CoE) and decreased need for mechanical ventilation (RR: 0.57; 95% CI: 0.42, 0.79; moderate CoE) [159].
In the study by Goldman et al that compared five and ten days of treatment, the shorter course of remdesivir showed a trend toward decreased mortality (RR: 0.75; 95% CI: 0.51, 1.12; low CoE) and increased clinical improvement at 14 days (RR: 1.19; 95% CI: 1.01, 1.40; low CoE); however, the evidence is uncertain because the persons in the 10-day group had more severe disease at baseline and there is the possibility of residual confounding despite the adjusted analysis [161].
Hospitalized patients on invasive ventilation and/or ECMO
Treatment with remdesivir failed to show a reduction in mortality (RR: 1.23; 95% CI: 0.99, 1.53; low CoE). Similarly, remdesivir failed to show or exclude a reduction in time to recovery among patients on invasive ventilation and/or ECMO (HR: 0.98; 95% CI: 0.70, 1.36; very low CoE).
Harms
Patients with mild-to-moderate disease who are at high risk for progression to severe COVID-19
As with other remdesivir studies published so far, three days of remdesivir infusions did not appear to be associated with a greater risk of serious adverse events compared to no remdesivir (RR: 0.27; 95% CI: 0.1, 0.7; moderate CoE).
Hospitalized patients with SpO2 ≤94% on room air
Patients treated with remdesivir do not appear to experience greater serious adverse events (grade 3/4) than those not receiving remdesivir (RR: 0.87; 95% CI: 0.59, 1.28; moderate CoE) [159, 160].
Patients receiving five days of remdesivir may experience fewer serious adverse events and adverse events leading to treatment discontinuation than patients receiving 10 days of remdesivir (RR: 0.61; 0.44, 0.85; low CoE and RR: 0.44; 95% CI: 0.21, 0.95; low CoE, respectively); however, this evidence is uncertain because of the increased severity of disease among patients in the 10-day arm [161].
Hospitalized patients on invasive ventilation and/or ECMO
Patients on invasive ventilation and/or ECMO treated with remdesivir do not appear to experience greater serious adverse events than those not receiving remdesivir (RR: 0.79; 95% CI: 0.54, 1.16; moderate CoE).
Other considerations
Patients with mild-to-moderate disease who are at high risk for progression to severe COVID-19
The panel agreed that the overall certainty of evidence for the treatment of patients with mild-to-moderate COVID-19 was low due to concerns about imprecision, as less than half of the original projected sample size was enrolled leading to few events and fragility of the effect estimate. However, compared to prior trials, giving remdesivir early in the course of the viral infection appears to have a robust effect within the limitation of a limited sample size. The panel agreed that benefits are likely to outweigh any potential harms in patients with COVID-19 who are at high risk for severe disease. The evidence confirms that using remdesivir early in the disease process when viral loads are high confers maximum benefit. It is critical to make a rapid diagnosis and treat ambulatory patients with COVID-19 early in the disease course.
Hospitalized patients with SpO2 ≤94% on room air
The panel agreed that the overall certainty of the evidence for treatment of persons with severe disease with remdesivir compared to no remdesivir treatment was moderate due to concerns with imprecision. Given the inconsistent definition used in the evidence to describe baseline severity, the panel recognized a knowledge gap when assessing whether greater benefit could be attained for patients with oxygen saturation >94% and no supplemental oxygen; however, they agreed that the reported data supported the prioritization of remdesivir among persons with severe but not critical COVID-19.
The panel agreed on the overall certainty of the evidence for treatment with a five-day course compared to a 10-day course of treatment as low due to concerns with risk of bias and imprecision. The panel recognized the benefit of a shorter course of treatment, if providing similar or greater efficacy, on the availability of remdesivir. However, in a subgroup analysis of mechanically ventilated patients, the duration of treatment was 10 days in ACCT-1 trial; therefore, the panel recognized that a longer course of treatment could be desirable in this population.
Hospitalized patients on invasive ventilation and/or ECMO
The panel agreed on the overall certainty of the evidence for treatment of patients on invasive ventilation and/or ECMO with remdesivir as very low due to concerns with risk of bias and imprecision. The panel recognized that the estimates of effect for mortality and time to recovery exclude almost any benefit.
Pediatric use
The evidence for the use of remdesivir in children is limited. For ambulatory children at risk for severe disease, the RCT included 8 children aged 12 to 18 years, limiting our confidence in the available direct evidence for ambulatory care.
There are no randomized controlled data assessing efficacy of remdesivir for treatment of hospitalized pediatric patients with COVID-19. A report of 77 children who received remdesivir through compassionate use early in the pandemic found good tolerability in this population with a low rate of serious adverse events [162].
An ongoing study of remdesivir in children [163] is using 5 mg/kg on day one (maximum dose 200 mg) followed by 2.5 mg/kg daily in patients over 14 days of age, gestational age more than 37 weeks, and weight greater than or equal to 2.5 kg. The FDA EUA applies to patients weighing over 3.5 kg and applies to the lyophilized powder formulation only.
Conclusions and research needs for this recommendation
The guideline panel suggests remdesivir for patients with mild-to-moderate disease who are at high risk for severe COVID-19.
The guideline panel suggests remdesivir rather than no remdesivir for treatment of severe COVID-19 in hospitalized patients with SpO2 <94% on room air. However, the guideline panel suggests against the routine initiation of remdesivir among patients on invasive ventilation and/or ECMO. Additional clinical trials are needed to provide increased certainty about the potential for both benefit and harms of treatment with remdesivir, as well as to understand the benefit of treatment based on disease severity.
Prescribing information in the United States recommends against use of remdesivir in patients with estimated glomerular filtration rate less than 30 mL per minute. This recommendation arises from concern about accumulation of the excipient (betadex sulfobutyl ether sodium) in such patients with potential for hepatic and renal toxicity due to that substance. Additional research into safety of remdesivir in patients with reduced renal function is needed to ascertain whether this concern is substantiated.
Immunocompromised patients who are unable to control viral replication may still benefit from remdesivir despite SpO2 that exceeds 94% on room air or a requirement for mechanical ventilation. Management of immunocompromised patients with uncontrolled viral replication is a knowledge gap and additional research into such populations is needed.
In addition, research is needed to address gaps in the evidence of effectiveness of remdesivir based on viral load.
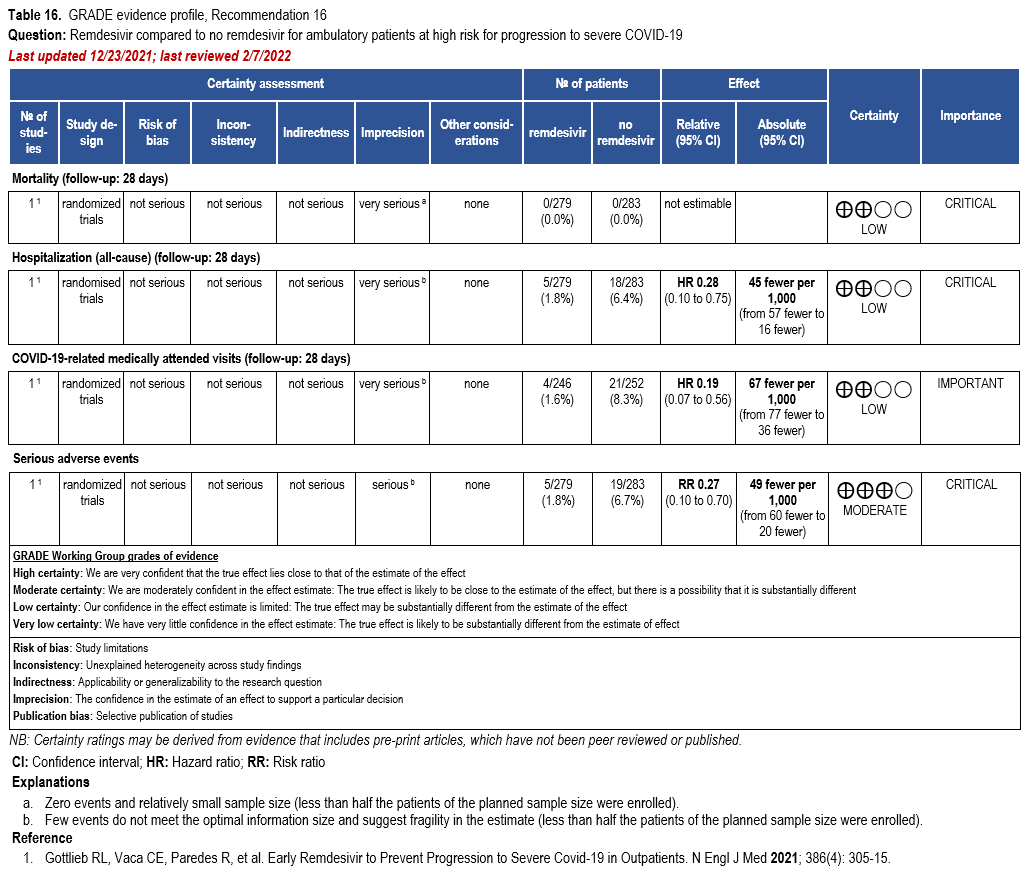
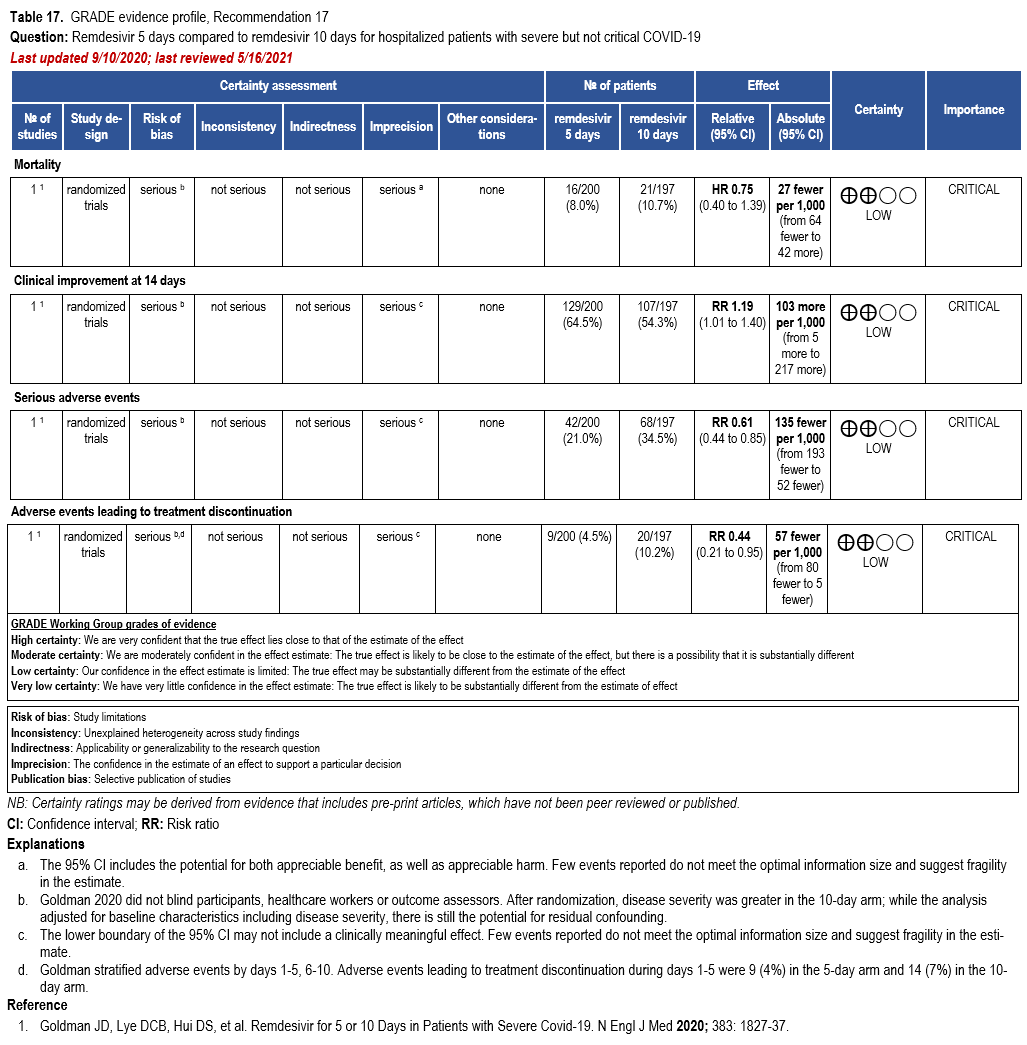
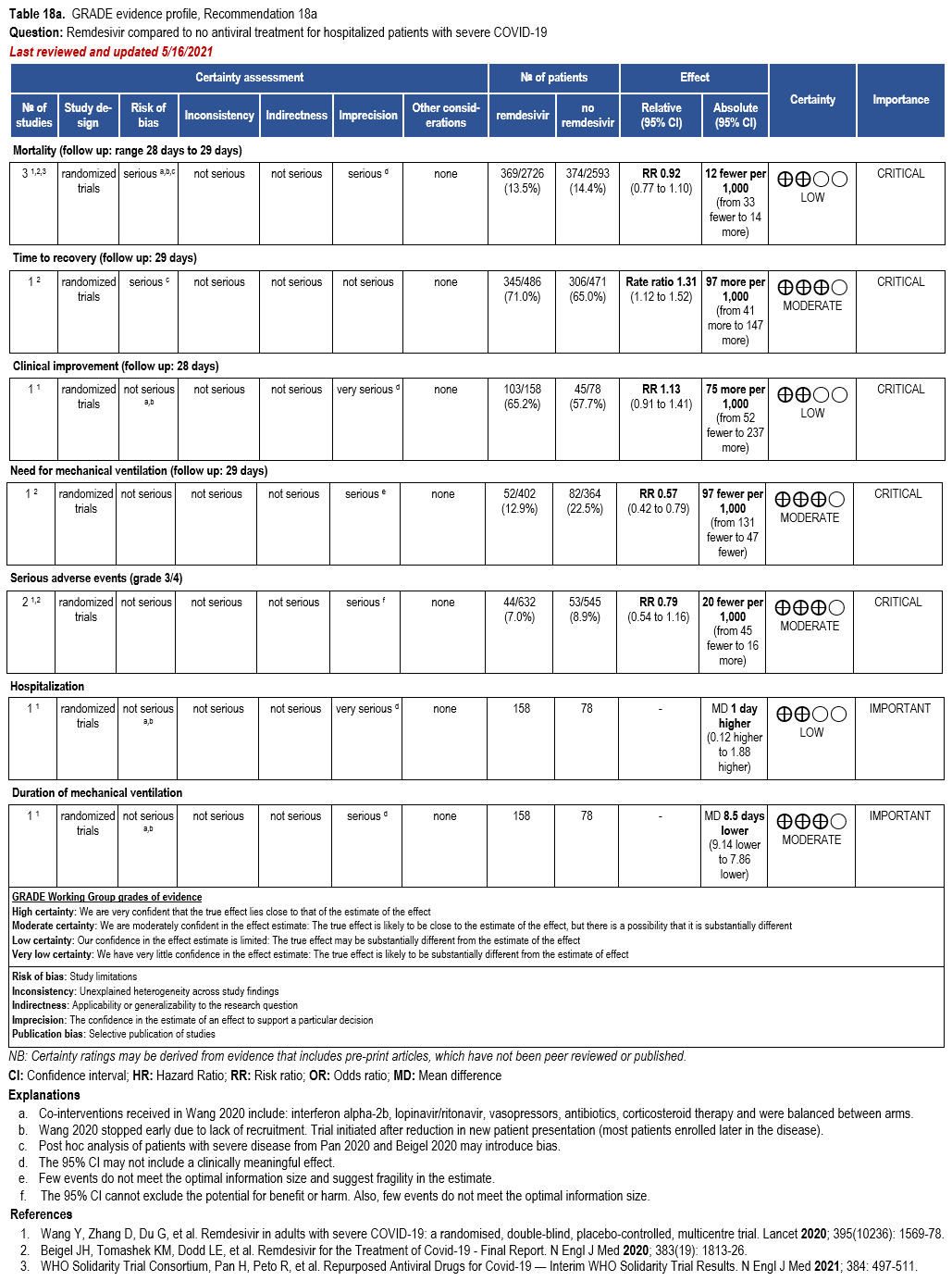
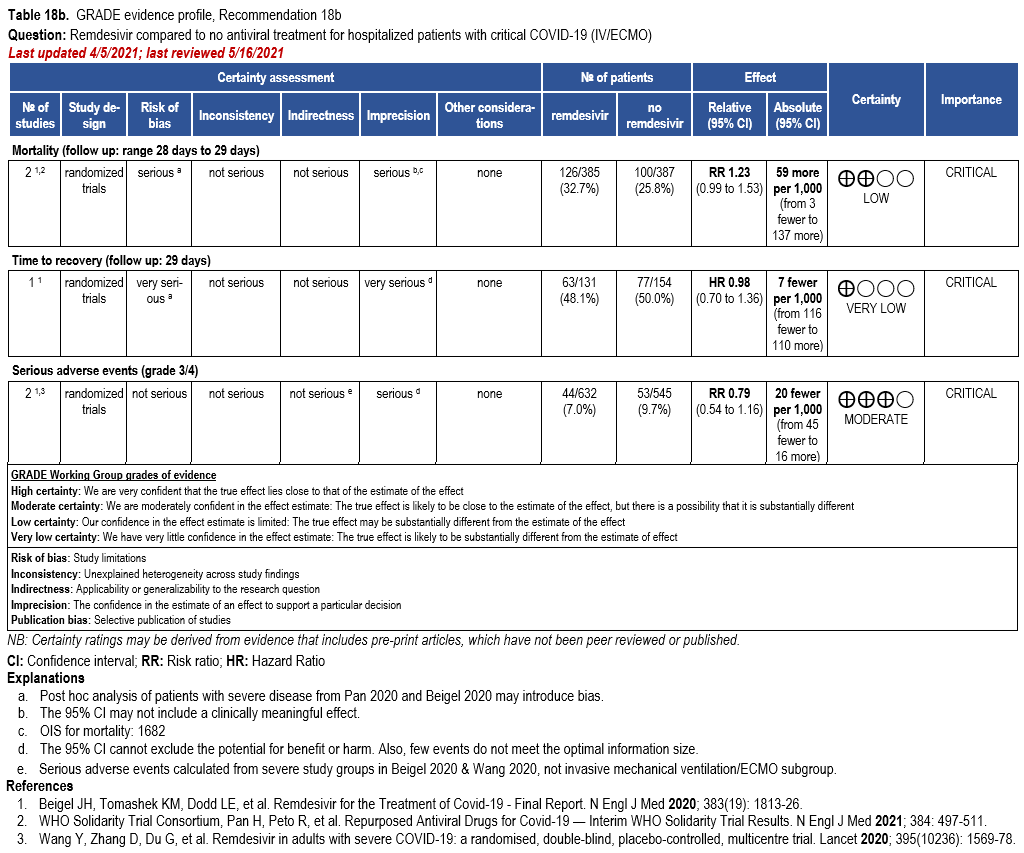
Supplementary Information
Study characteristics:
- Table s17. Remdesivir vs. no remdesivir (hospitalized patients)
- Table s18. Remdesivir vs. no remdesivir (ambulatory patients)
Forest plots:
- Figure s8a. Outcome of mortality for remdesivir vs. no remdesivir in hospitalized patients with severe disease
- Figure s8b. Outcome of severe adverse events (grade 3/4) for remdesivir vs. no remdesivir in hospitalized patients with severe disease
- Figure s8c. Outcome of mortality for remdesivir vs. no remdesivir in hospitalized patients on invasive ventilation and/or ECMO
- Figure s8d. Outcome of serious adverse events (grade 3/4) for remdesivir vs. no remdesivir in hospitalized patients on invasive ventilation and/or ECMO
Risk of bias:
Recommendations 19-20: Famotidine
Section last reviewed and updated 5/23/2022
Last literature search conducted 4/30/2022
Recommendation 19: Among ambulatory patients with mild-to-moderate COVID-19, the IDSA panel suggests against famotidine for the treatment of COVID-19 (Conditional recommendation††, Low certainty of evidence)
Recommendation 20: Among hospitalized patients with severe* COVID-19, the IDSA panel suggests against famotidine for the treatment of COVID-19. (Conditional recommendation††, Low certainty of evidence)
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is famotidine considered for treatment?
Anecdotal reports from China and a cohort study from the United States had suggested that patients infected with SARS-CoV-2 who were receiving famotidine, an H2-receptor antago-nist used for conditions such as gastroesophageal reflux and peptic ulcer disease, had improved survival versus those receiving proton pump inhibitors (PPIs) [164, 165]. This study led to inter-est in the drug, though no predominant theory describing a mechanism for its efficacy yet exists.
Our search identified two RCTs comparing treatment with famotidine against no famotidine among ambulatory persons with COVID-19 and persons hospitalized with severe COVID-19 [166, 167] (Table 19-20).
Summary of the evidence
Ambulatory patients with mild-to-moderate disease
One patient and assessor blinded RCT examined high-dose famotidine at 80 mg three times daily for 14 days (n=27) vs placebo (n=28) in a predominantly younger population (35 years of age) at average risk for progression to severe disease [166]. Symptom resolution was the primary endpoint.
Hospitalized patients with severe disease
Oral famotidine at standard doses of 40 mg daily (n=89) vs placebo (n=89) was given to hospitalized patients with severe COVID-19 in an open-label RCT. The authors recorded symptom resolution, length of hospital stay, need for ICU care, need for mechanical ventilation, or death [167].
Benefits
Ambulatory patients with mild-to-moderate disease
Symptom resolution in ambulatory patients at day 28 failed to show or to exclude a beneficial effect of high-dose famotidine (RR: 1.1, 95% CI: 0.76, 1.58 – not directly reported but estimated from the survival curve; low CoE).
Hospitalized patients with severe disease
In hospitalized patients with severe COVID-19, famotidine at standard dose failed to show or exclude a beneficial effect on mortality, need for mechanical ventilation, or need for ICU care (RR: 0.89, 95% CI: 0.36, 2.2; RR: 0.88, 95% CI: 0.53, 1.45; RR: 0.9, 95% CI: 0.51, 1.58, respectively; all low CoE). Time to symptom resolution was shorter in the famotidine group (MD -0.9 days, 95% CI: -1.44, -0.36), as was length of hospital stay (MD -1.7 days, 95% CI: -2.77, -1.13), although due to lack of blinding these estimates remain less certain (low CoE) (Table 20).
Harms
At standard doses, famotidine is well tolerated. Common adverse events include diarrhea or constipation but occur in less than 5% of people. Severe adverse events occur in less than 1% of persons taking famotidine. Adverse events were rare in the ambulatory study examining high dose famotidine (RR: 0.69, 95% CI: 0.13, 3.8) and no severe adverse events were reported.
Other considerations
The panel determined the certainty of evidence for ambulatory patients with mild-to-moderate disease to be low due to concerns with imprecision due to small sample sizes and few events.
The panel determined the certainty of evidence for hospitalized patients with severe disease to be low due to concerns with risk of bias and imprecision from small sample sizes and few events.
Conclusions and research needs for this recommendation
The guideline panel suggests against famotidine for the sole purpose of treating COVID-19. Clinical trials with larger sample sized would be needed to determine the true effect of famotidine in patients with COVID-19 (Supplementary Table s2).
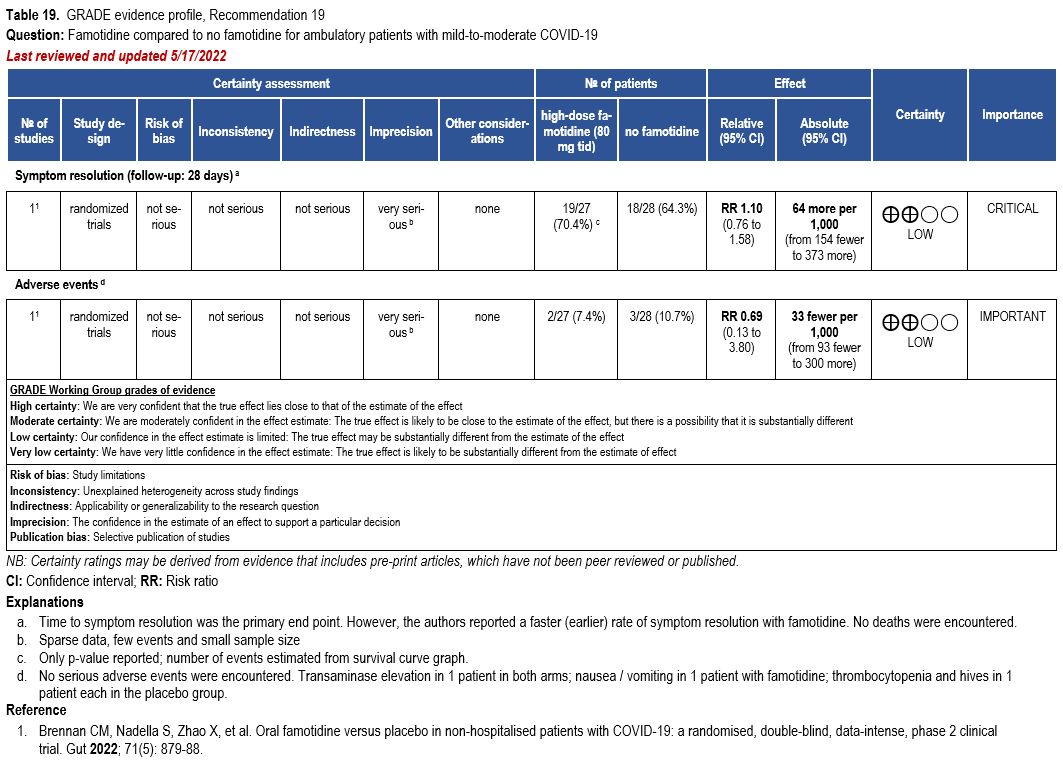
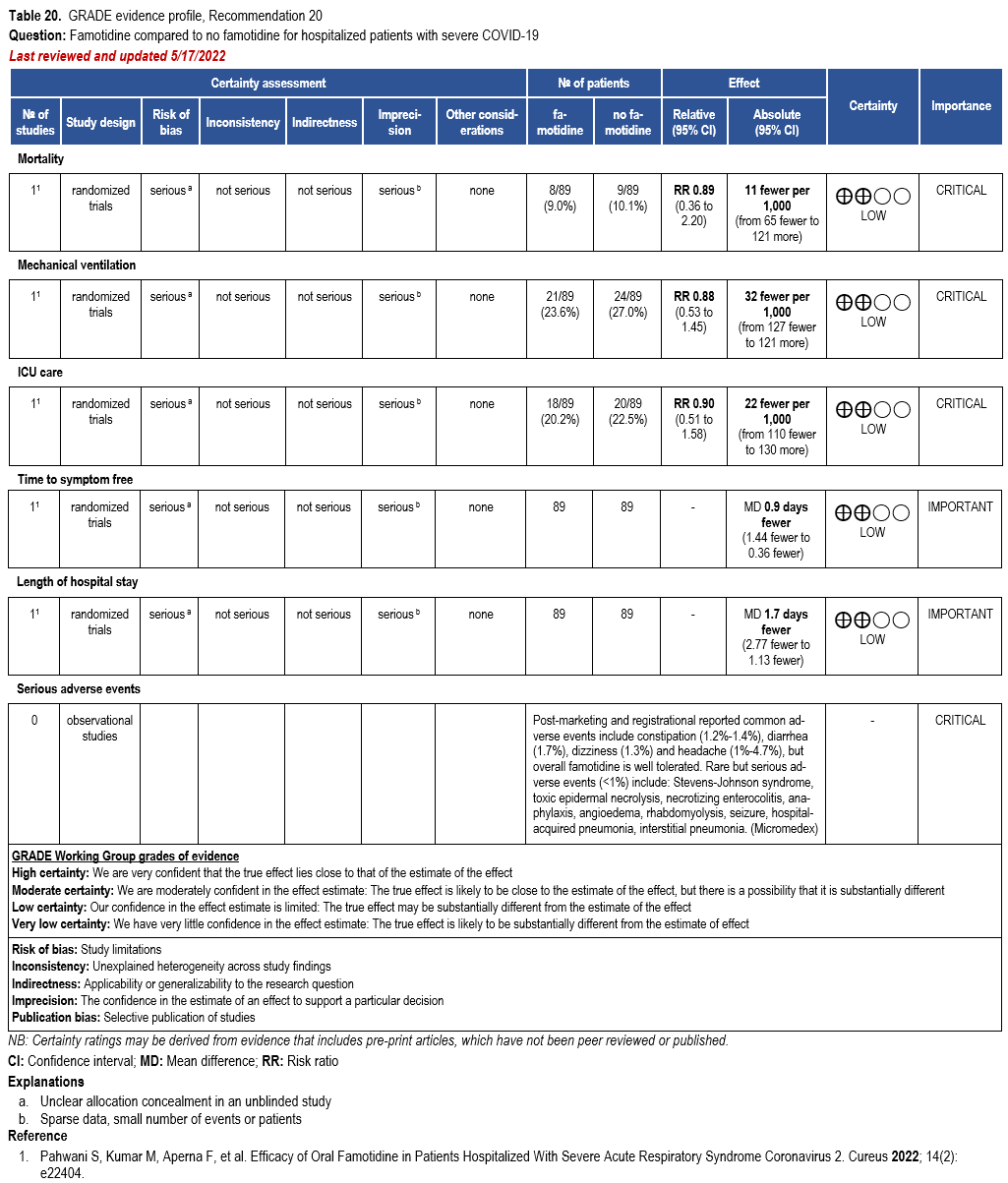
Supplementary Information
Study characteristics:
Risk of bias:
Neutralizing antibodies for pre-exposure prophylaxis
Section last reviewed and updated on 1/27/2023
As of 1/26/2023, based on CDC Nowcast data, fewer than 10% of circulating variants in the US are susceptible to tixagevimab/cilgavimab (Evusheld), the sole product that has been available for pre-exposure prophylaxis. Tixagevimab/cilgavimab is therefore no longer authorized for use in the US until further notice by FDA.
SARS-CoV-2 is expected to continue to evolve. Although the general trend has been towards increasing resistance to neutralizing monoclonal antibodies, there have been instances in which new variants became more susceptible to existing anti-SARS CoV-2 neutralizing antibodies. Should this occur again, or should newly developed, more active neutralizing antibodies be authorized for prophylaxis, the panel will offer recommendations regarding use.
Please see the retired versions of this section below:
Neutralizing antibodies for post-exposure prophylaxis
Section last reviewed and updated on 1/12/2023
As the pandemic progressed, new SARS CoV-2 variants emerged with reduced susceptibility to various anti-SARS-CoV-2 neutralizing antibodies in assays performed using infectious (also referred to as authentic) and pseudotyped viruses. The first two US FDA authorized anti-SARS-CoV-2 neutralizing antibody combinations, bamlanivimab/etesevimab and casirivimab/imdevimab, were found to be largely inactive against the Omicron BA.1 and BA.2 variants, rendering these products no longer useful for either treatment or post-exposure prophylaxis. As a result, Emergency Use Authorization was withdrawn by the US FDA for both bamlanivimab/etesevimab and casirivimab/imdevimab, leaving no available neutralizing antibody product for use in the United States for post-exposure prophylaxis. Should new variants become susceptible to an existing neutralizing antibody or should newly developed, more susceptible neutralizing antibodies be authorized for post-exposure prophylaxis, the panel will offer recommendations regarding use.
For areas of the world where a significant proportion of circulating variants retain susceptibility to at least one neutralizing antibody authorized for post-exposure prophylaxis, use could be considered. However, data are scarce on how susceptibility reductions affect clinical efficacy, relative to that observed prior to emergence of novel variants.
Neutralizing antibodies for treatment
Section last reviewed and updated 1/12/2023
During 2022, multiple Omicron sub-variants with progressively greater in vitro reductions in susceptibility to multiple anti-SARS CoV-2 neutralizing antibodies emerged. On November 30, 2022, the US FDA withdrew Emergency Use Authorization for bebtelovimab, the one anti-SARS CoV-2 neutralizing antibody product that had retained in vitro activity against most previously circulating SARS-CoV-2 variants, leaving no available neutralizing antibody product in the United States for treatment of COVID-19.
For areas of the world where a significant proportion of circulating variants retain susceptibility to at least one authorized therapeutic neutralizing antibody, use could be considered, taking into account the predicted relative benefits of the anti-SARS CoV-2 neutralizing antibody product compared with alternative antiviral therapies. However, data are scarce on how susceptibility reductions affect clinical efficacy, relative to that observed prior to emergence of novel variants.
SARS-CoV-2 is expected to continue to evolve. Although the general trend has been towards increasing resistance to therapeutic neutralizing monoclonal antibodies, there have been instances in which new variants became more susceptible to existing anti-SARS CoV-2 neutralizing antibodies. Should this occur again, or should newly developed, more active neutralizing antibodies be authorized for treatment, the panel will offer recommendations regarding use.
Please see the retired version of this section below:
Recommendations 21-23: Janus kinase inhibitors (baricitinib and tofacitinib)
Baricitinib
Section last reviewed and updated 4/29/2022
Last literature search conducted 3/31/2022
Recommendation 21: Among hospitalized adults with severe* COVID-19, the IDSA panel suggests baricitinib with corticosteroids rather than no baricitinib. (Conditional recommendation†, Moderate certainty of evidence)
Remarks:
- Baricitinib 4 mg per day (or appropriate renal dosing) up to 14 days or until discharge from hospital.
- Baricitinib appears to demonstrate the most benefit in those with severe COVID-19 on high-flow oxygen/non-invasive ventilation at baseline.
- Limited additional data suggest a mortality reduction even among patients requiring mechanical ventilation.
Recommendation 22: Among hospitalized patients with severe* COVID-19 who cannot receive a corticosteroid (which is standard of care) because of a contraindication, the IDSA guideline panel suggests use of baricitinib with remdesivir rather than remdesivir alone. (Conditional recommendation†, Low certainty of evidence)
- Remark: Baricitinib 4 mg daily dose for 14 days or until hospital discharge. The benefits of baricitinib plus remdesivir for persons on mechanical ventilation are uncertain.
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen, oxygen through a high-flow device, or non-invasive ventilation.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is baricitinib considered for treatment?
Baricitinib, a selective Janus kinase 1 and 2 (JAK1 and JAK2, respectively) inhibitor currently FDA-approved for the treatment of RA, is being investigated in multiple studies for treatment of COVID-19. The proposed benefits of baricitinib in the management of COVID-19 may be two-fold as it has both anti-inflammatory and potential antiviral activity [168]. Janus kinase (JAK) mediates cytokine signaling, which contributes to inflammation; JAK inhibitors, therefore, may decrease cytokine-mediated inflammation. Baricitinib inhibits host intracellular membrane proteins AP2-associated protein kinase 1 (AAK1) and also binds cyclin G-associated kinase (GAK), both thought to play a role in receptor mediated endocytosis of many viruses including Ebola, dengue, hepatitis C, and SARS-CoV-2 [169-171]. Baricitinib has been evaluated in people with COVID-19 in both randomized and non-randomized studies [172-176].
Based on experience in clinical trials for RA, baricitinib has been associated with an increased risk of adverse effects including infections (especially upper respiratory tract infections), thrombosis, lymphopenia, anemia, increases in lipids, elevations in liver enzymes, and elevations in creatinine phosphokinase [168]. In clinical trials for RA, baricitinib was associated with a numerically higher risk of upper respiratory tract infections and herpes simplex and herpes zoster infections compared with placebo [177]. Opportunistic infections such as herpes simplex, herpes zoster, and tuberculosis [178, 179] have been reported in patients taking baricitinib. Many of these side effects appear to be dose related, with increased incidence in patients taking baricitinib 4 mg compared with 2 mg. Patients enrolled in Adaptive COVID-19 Treatment Trial (ACTT-2), COV-BARRIER and RECOVERY (Randomized evaluation of COVID-19 Therapy) received baricitinib 4 mg daily for 2-14 days or until discharge, a shorter duration than those taking the drug for RA.
Patients with COVID-19 have been found to have abnormalities in coagulation parameters and might have an elevated risk of thrombosis [180]. Baricitinib receipt was associated with an increased incidence of thrombosis when compared with placebo receipt in clinical trials for its FDA approval for RA, especially at a higher dose of 4 mg daily [168]. During the 16-week treatment period in RA trials, venous thromboembolism (VTE) occurred in five patients treated with baricitinib 4 mg daily, compared with zero in the 2 mg daily and placebo groups. Arterial thrombosis occurred in two patients treated with baricitinib 4 mg, two patients treated with baricitinib 2 mg, and one patient on placebo. In ACTT-2, the percentage of patients reported to have VTE was numerically higher in the combination group (21 patients [4.1%] vs. 16 patients [3.1%]) although it was similar overall (absolute difference 1%, 95% CI -1.3 to 3.3) [181]. Of note, all patients in ACTT-2 were recommended to receive VTE prophylaxis if they had no contraindication. We do not have long-term data, especially on safety, development of the aforementioned adverse effects, and opportunistic infections from these two trials.
Summary of the evidence
Baricitinib
Our literature search identified two randomized controlled trials (RCTs) that compared the use of baricitinib (4 mg daily dose up to 14 days) to placebo in hospitalized adults. One trial, COV-BARRIER, included patients with severe COVID (NIAID OS: 4 – hospitalized, not requiring supplemental oxygen; 5 – hospitalized, requiring supplemental oxygen; or 6 – hospitalized, receiving non-invasive ventilation or high-flow oxygen devices) [176, 182, 183]. Critically ill and mechanically ventilated patients (OS7) were excluded from COV-BARRIER study. In the COV-BARRIER trial, randomization was stratified by disease severity, age, region, and use of corticosteroids. Participants in both arms had >1 elevated inflammatory marker (CRP, d-dimer, lactate dehydrogenase, ferritin) and also received standard of care, which included corticosteroids in 79% and/or antivirals (e.g., remdesivir in 18.9%). The RECOVERY, trial included patients hospitalized for COVID-19. Approximately, 70% of patients received supplemental oxygen, 25% received non-invasive ventilation, and 3% received invasive ventilation. Participants in both arms received standard of care, which included corticosteroids in approximately 95% and/or antivirals (e.g., remdesivir in 20%).
An additional exploratory trial subsequent to the COV-BARRIER primary trial of baricitinib treatment for critically ill (OS-7) patients with COVID-19 pneumonia requiring invasive mechanical ventilation was identified that reported on the outcomes of mortality, need for invasive mechanical ventilation, days of hospitalization, and serious adverse events [184].
Baricitinib without corticosteroids, with remdesivir
Our literature search identified one RCT that reported on the use of baricitinib (4 mg daily dose) plus remdesivir in hospitalized patients with moderate and severe COVID-19 ([181]. This trial was conducted as the second stage of the ACTT-2, where subjects were randomized to receive combination therapy with baricitinib and remdesivir or remdesivir alone [181] (Table 23). Randomization was stratified by disease severity classified by an OS of clinical status (4+5 vs 6+7 [7 –patients with an ordinal scale of 6 (high-flow oxygen and non-invasive ventilation) or 7 (mechanical ventilation or ECMO). Mild-to-moderate disease was defined as patients with an ordinal scale of 4 (hospitalized, but not requiring supplemental oxygen) or 5 (requiring supplemental oxygen). The trial was initiated before corticosteroids were commonly used for severe COVID-19.
Benefits
Baricitinib
Treatment of hospitalized patients with severe COVID-19 with baricitinib rather than no baricitinib reduced 60-day mortality (RR 0.87; 95% CI: 0.78 to 0.96; moderate CoE). The odds of COVID-19 disease progression trends toward a reduction in persons receiving treatment with baricitinib (OR: 0.85; 95% CI: 0.67, 1.08; moderate CoE), as well as the risk of needing mechanical ventilation (RR: 0.85; 95% CI: 0.73, 0.99; moderate CoE).
Treatment of critically ill hospitalized patients with baricitinib rather than no baricitinib reduced the risk of 60-day mortality (RR 0.74; 95% CI: 0.57 to 0.97; moderate CoE).
Baricitinib without corticosteroids, with remdesivir
In ACTT-2, the combination of baricitinib and remdesivir showed a trend towards lower mortality (4.7% vs. 7.1%; rate ratio: 0.65; 95% CI 0.39, 1.09; moderate CoE). In patients stratified within the severe COVID-19 pneumonia group, defined as 6 or 7 on the ordinal scale, subjects who received baricitinib and remdesivir were more likely to experience clinical recovery (defined as a value of <4 on the ordinal scale) at day 28 (69.3% vs. 59.7%; rate ratio 1.29; 95% CI 1.00, 1.66; moderate CoE). The original stratification was altered as 40 subjects were misclassified at baseline; however, re-analysis of the original stratified data produced a similar result. Patients in the baricitinib arm were less likely to require initiation of mechanical ventilation or ECMO through day 29 (10% vs. 15.2%; RR: 0.66; 95% CI 0.46, 0.93; low CoE). In summary, it appeared that patients requiring supplemental oxygen or non-invasive ventilation at baseline benefitted most from baricitinib; the benefit was less clear in patients already on mechanical ventilation.
Harms
The risk of serious adverse events in hospitalized patients with severe or critical COVID-19 receiving baricitinib was not greater than those not receiving baricitinib (RR: 0.82; 95% CI: 0.65, 1.03; moderate CoE and RR 0.70; 95% CI: 0.50 to 0.97, moderate CoE, respectively). Patients who were immunocompromised (i.e., received immunosuppressant drugs or were neutropenic) and had a history of recent of thromboembolism were not excluded from the RECOVERY trial, unlike BARRIER-COV trial. Non-comparative serious adverse events were reported in the RECOVERY 2022 trial (baricitinib N=4,148): 13 total (5 serious infections, 3 bowel perforations, 2 pulmonary embolisms, 1 each of ischemic colitis, elevated transaminases and seizure).
In ACTT-2, patients receiving baricitinib and remdesivir had a lower risk of developing any serious adverse events through day 28 (16% vs. 21%; RR 0.76; 95% CI 0.59, 0.99; moderate CoE) whether or not thought to be related to the study drug. In this trial, the overall rate of new infections was lower in the baricitinib plus remdesivir group compared with remdesivir alone (30 patients [5.9%] versus 57 patients [11.2%]) [181]. However, patients who received concomitant glucocorticoids had a higher incidence of serious or non-serious infections as compared with those who did not: 25.1% and 5.5%, respectively. It was not specified what proportion of these patients in the study were in the baricitinib combination group versus the control group.
Other considerations
Baricitinib
The panel agreed on the overall certainty of evidence as moderate due to concerns with imprecision, as some outcomes have concerns with fragility. The guideline panel recognized the resource implications based on the dose and duration reported in the trial (4 mg daily up to 14 days). Additional data from hospitalized patients with critical COVID-19 suggest consistent benefits; however, there are concerns with imprecision based on a small sample in this group.
Baricitinib without corticosteroids
The panel agreed that the overall certainty of evidence was low due to concerns with risk of bias, driven by the use of data from post hoc analyses and imprecision, which recognized the limited events and concerns with fragility in the group who likely benefited most (those requiring supplemental oxygen or non-invasive ventilation). The guideline panel noted the importance of suggesting baricitinib plus remdesivir as an option for persons unable to receive corticosteroids.
Conclusions and research needs for this recommendation
The guideline panel suggests baricitinib in addition to standard of care for patients hospitalized with severe COVID-19. The guideline panel suggests baricitinib with remdesivir for persons for whom corticosteroids are indicated but who cannot receive them due to a contraindication. Baricitinib plus remdesivir should be reserved for patients who cannot take corticosteroids because dexamethasone has been proven to reduce mortality in patients hospitalized with COVID-19 who require supplemental oxygen or mechanical ventilation and, for this reason, dexamethasone is recommended by the panel for this group. It is uncertain whether baricitinib plus remdesivir will have the same benefit as dexamethasone. As of the time of this narrative, there are no head-to-head trials evaluating either the combination of baricitinib plus tocilizumab or evaluating baricitinib compared to tocilizumab. A post hoc subgroup analysis in the RECOVERY trial showed no difference in measured outcomes with concomitant baricitinib and tocilizumab, but further well-done studies are needed [200].

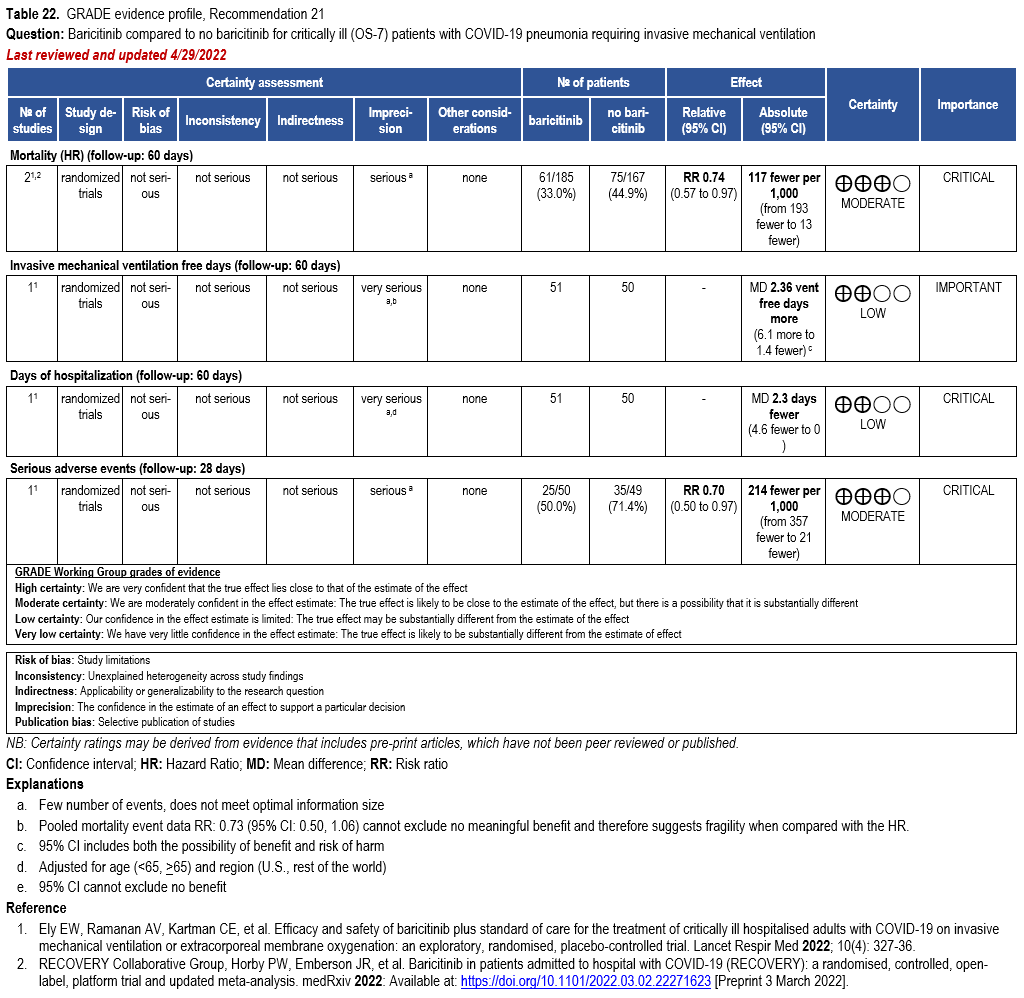
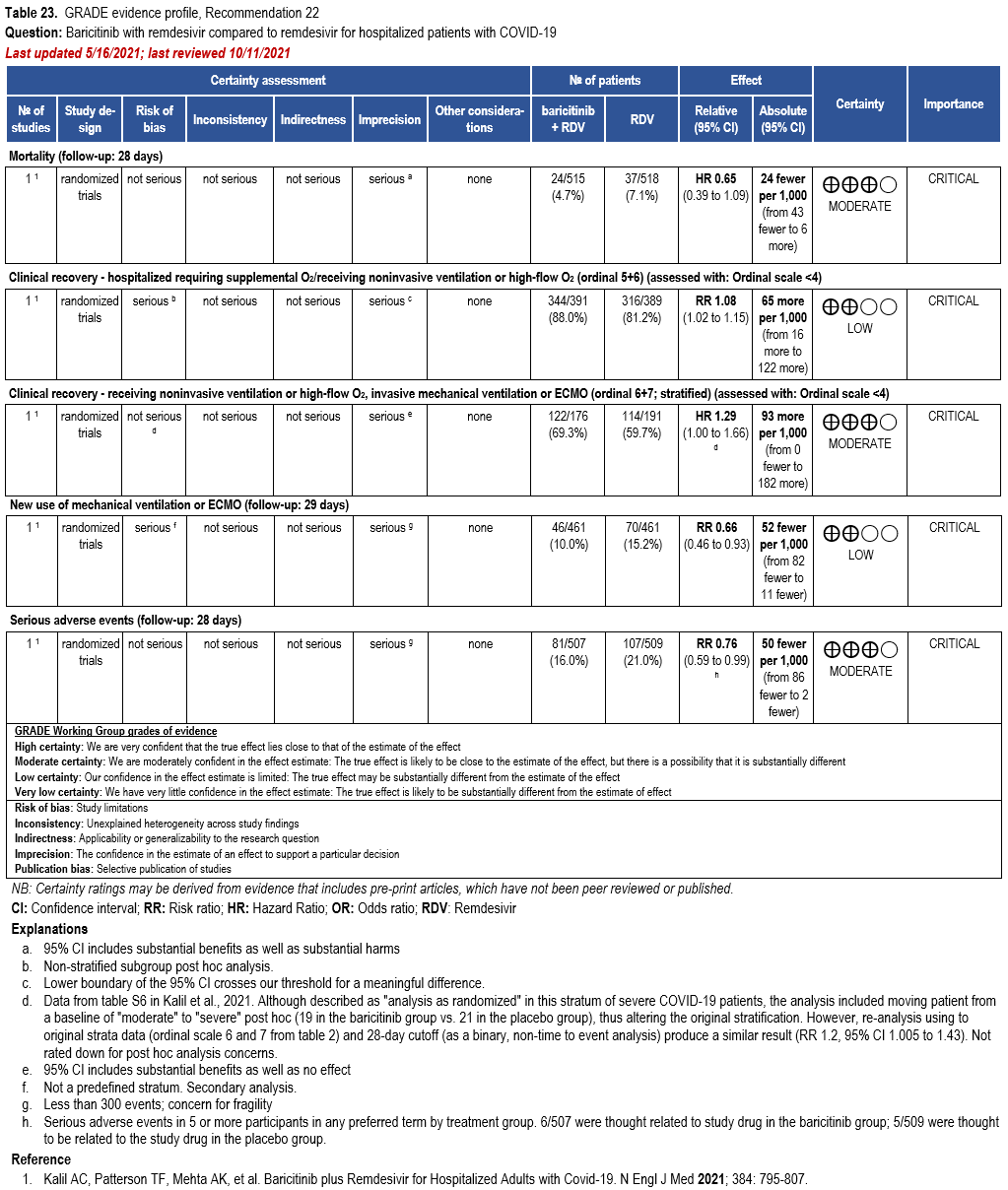
Supplementary Information
Study characteristics:
Risk of bias:
Tofacitinib
Section last reviewed and updated 8/21/2021
Last literature search conducted 7/31/2021
Recommendation 23: Among hospitalized adults with severe* COVID-19 but not on non-invasive or invasive mechanical ventilation, the IDSA panel suggests tofacitinib rather than no tofacitinib. (Conditional recommendation†, Low certainty of evidence)
Remarks:
- Tofacitinib appears to demonstrate the most benefit in those with severe COVID-19 on supplemental or high-flow oxygen.
- Patients treated with tofacitinib should be on at least prophylactic dose anticoagulant.
- Patients who receive tofacitinib should not receive tocilizumab or other IL-6 inhibitor for treatment of COVID-19.
- The STOP-COVID Trial did not include immunocompromised patients.
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen or oxygen through a high-flow device.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is tofacitinib considered for treatment?
Tofacitinib is a JAK inhibitor that preferentially inhibits JAK-1 and JAK-3 though it is active on all other JAK isoforms. It is FDA-approved for moderate to severe RA, active psoriatic arthritis, and moderate to severe ulcerative colitis. Like baricitinib, it is expected that JAK inhibition leads to downstream suppression of cytokine production, thereby modulating the inflammatory cascade that results in systemic inflammation in patients with severe COVID-19. See baricitinib section (above) for additional rationale on considerations for treatment.
Summary of the evidence
Our literature search identified one RCT that compared the use of tofacitinib 10 mg every 12 hours for up to 14 days or placebo [185]. Patients included were those who had laboratory-confirmed SARS-CoV-2 infection and evidence of COVID-19 pneumonia on imaging and who were hospitalized for less than 72 hours. Patients in this study could not be receiving non-invasive ventilation, mechanical ventilation, or ECMO at baseline. Additionally, patients with a history of or current thrombosis, personal or first-degree family history of blood clotting disorders, immunosuppression, any active cancer, or those with certain cytopenias were excluded from this trial. Patients who received other potent immunosuppressants, or other biologic agents were excluded, while the use of glucocorticoids for the management of COVID-19 was permitted. A composite outcome of death at day 28 or respiratory failure (defined as progression to NIAID ordinal scale 6, 7, or 8) was the primary outcome.
Benefits
Treatment of hospitalized patients with COVID-19 pneumonia with tofacitinib resulted in a lower risk of the composite outcome of death or respiratory failure compared to no tofacitinib (RR: 0.63; 95% CI: 0.41, 0.97; low CoE). However, results failed to show or to exclude a beneficial or detrimental effect on mortality alone (RR: 0.49; 95% CI: 0.15, 1.63; low CoE) or progression to mechanical ventilation or ECMO by day 28 (RR: 0.25; 95% CI: 0.03, 2.20; low CoE).
Harms
Patients who received tofacitinib experienced more serious adverse events; however, this may not be meaningfully different from those that received placebo (RR: 1.18; 95%CI: 0.64, 2.15; low CoE). Use of tofacitinib for other indications has shown an increase in thrombotic events which prompted a black box warning by the FDA [186, 187]. As COVID-19 infection itself increases the risk for VTE events; it is important to note that the patients studied were either on prophylactic or full dose anticoagulation during treatment with tofacitinib.
Tofacitinib carries four black boxed warnings for its labeled indications including a warning for 1) serious infections including tuberculosis, invasive fungal infections, bacterial, viral and other opportunistic pathogens; 2) mortality; 3) thrombosis; and 4) lymphoma and other malignancies, including an increased rate of EBV-mediated post-transplant lymphoproliferative disorder [186-189].
Other considerations
The panel agreed that the overall certainty of evidence was low due to concerns of imprecision, which recognized the limited number of events and concerns about fragility of the results in the group who likely would benefit the most (those requiring supplemental oxygen or oxygen through a high-flow device).
Conclusions and research needs for this recommendation
The guideline panel suggests tofacitinib in addition to standard of care for patient hospitalized for severe COVID-19. Due to the increased risk of VTE with treatment with tofacitinib, patients should receive at least prophylactic doses of anticoagulants during their hospital stay. Patients who received JAK inhibitors should not receive tocilizumab or other immunomodulators as no adequate evidence is available for its combined use.
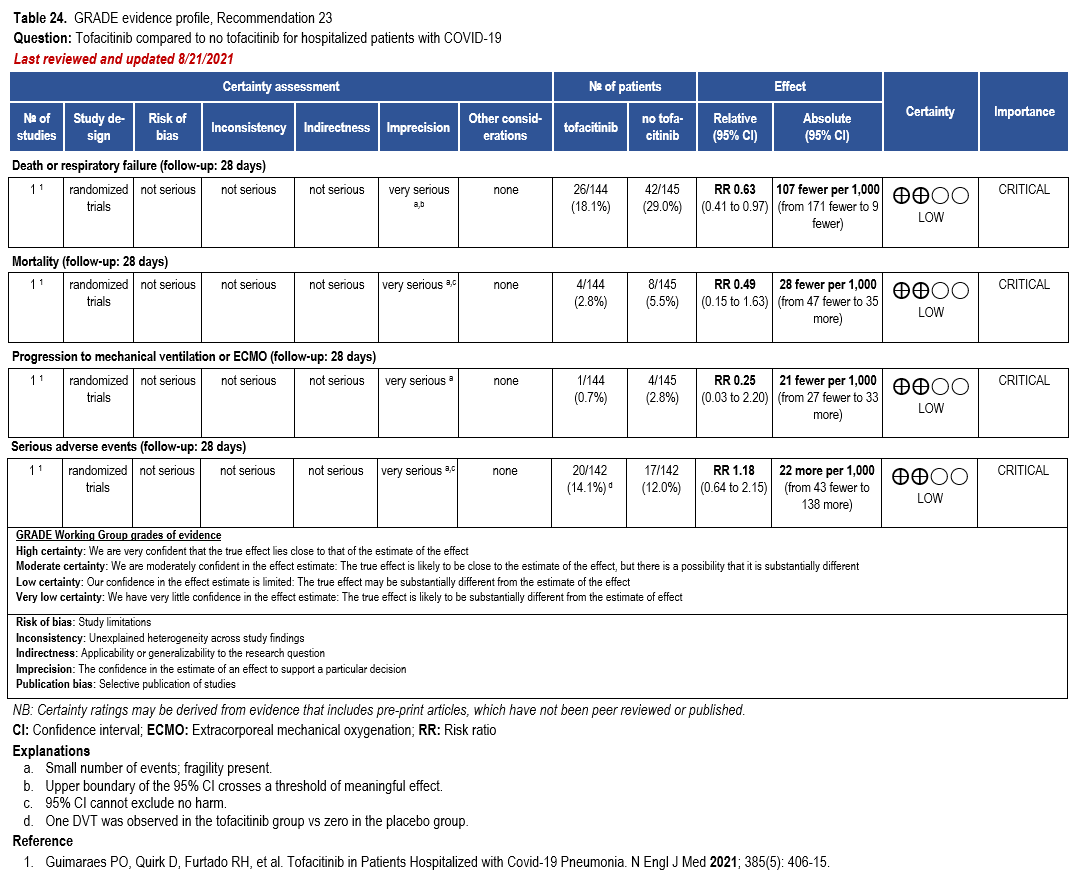
Supplementary Information
Study characteristics:
Risk of bias:
Recommendations 24-25: Ivermectin
Section last reviewed and updated 10/10/2022
Last literature search conducted 8/31/2022
Recommendation 24: In hospitalized patients with COVID-19, the IDSA panel suggests against ivermectin. (Conditional recommendation††, Very low certainty of evidence)
Recommendation 25: In ambulatory persons with COVID-19, the IDSA panel recommends against ivermectin. (Strong recommendation, Moderate certainty of evidence)
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is ivermectin considered for treatment?
Ivermectin is an anti-parasitic agent that is FDA-approved for onchocerciasis and strongyloidiasis and is used off-label for the treatment of many parasitic infections. Although it has in vitro activity against some viruses, including SARS-CoV-2, it has no proven therapeutic utility. In vitro activity against SARS-CoV-2 [190] requires concentrations considerably higher than those achieved in human plasma and lung tissue to reach the in vitro IC50 [191]. Ivermectin has been shown to have anti-inflammatory effects in in vitro and in vivo studies hence hypothesized to have a mechanism beyond its anti-viral effects in the treatment of COVID-19 [192, 193].
Since ivermectin is generally well tolerated, it was empirically evaluated in uncontrolled studies for COVID-19, alone and in combination with other off-label medications.
Summary of the evidence
Our search identified 28 studies in patients with COVID-19 with ages ranging between 8 and 86 years that reported on the outcomes of mortality, symptom resolution, viral clearance, and adverse events, and informed the evidence review for inpatient and outpatient therapy [194-214]. Eligible studies compared treatment with ivermectin against a placebo or standard of care. Studies comparing ivermectin to a non-placebo, active comparison (i.e., a different agent considered a possible treatment for COVID-19 infection by clinicians) or that did not provide a comparison arm were not included in these analyses. Several studies did not meet eligibility for inclusion in this review. Three trials compared ivermectin to hydroxychloroquine (comparison to treatment with evidence of harm) [215-217]; two trials examined ivermectin as prophylactic treatment [218, 219]; and two trials did not provide study data in a peer-reviewed, published, or pre-print manuscript [217, 220].
The studies that informed the recommendations for hospitalized patients included 15 randomized control trials (RCTs) [194-198, 202-205, 213, 214, 221-224]. Sixteen RCTs [196, 197, 199-201, 206-212, 224-227] informed the recommendation for ambulatory persons. Each of them compared an active treatment arm of ivermectin to an inactive comparison (e.g., standard of care with or without placebo).
The evidence informing the recommendations for treating hospitalized and ambulatory persons with ivermectin reported on the use of a range of doses (100 mcg/kg/day to 400 mcg/kg/day) and durations (one day up to seven days). Among studies reporting on hospitalized patients, substantial heterogeneity was observed, introduced by one study (Supplementary Table s9c) [194]. Ahmed 2020 treated patients with ivermectin for a duration of five days, rather than one day as used by the remaining studies. This may explain the heterogeneity between studies; however, excluding Ahmed 2020, any meaningful reduction in viral clearance was still not demonstrated by the summary estimate (Supplementary Table s9d). Heterogeneity was not observed for other outcomes reported for hospitalized or ambulatory persons.
Among the RCTs, the risk of bias was high in two trials because of unsuccessful randomization into treatment and control groups. Hashim et al (2020) [197] inadequately randomized participants by allocating them to respective treatment arms on odd and even days, as well as assigning all critically ill patients to the ivermectin arm, and Podder et al (2020) [198] allocated participants based on odd or even registration numbers. In addition, across many RCTs, there were concerns due to lack of blinding of study personnel, which may lead to over- or under-estimates of treatment effects, particularly for subjective outcomes (e.g., symptom resolution, adverse events).
Benefits
Hospitalized
The evidence from RCTs failed to demonstrate a meaningful effect on mortality or need for mechanical ventilation among persons with COVID-19 (risk ratio [RR]: 0.85; 95% confidence interval [CI]: 0.40, 1.84; moderate certainty of evidence [CoE] and RR: 0.45; 95% CI: 0.24, 0.86, low CoE, respectively). Persons receiving treatment with ivermectin rather than no ivermectin failed to demonstrate a beneficial or detrimental effect on symptom resolution or viral clearance at day seven (RR: 1.07; 95% CI: 0.69, 1.65; very low CoE and RR: 1.06; 95% CI: 0.74, 1.52; very low CoE, respectively).
Ambulatory
Treatment with ivermectin does not reduce mortality (RR: 0.86; 95% CI: 0.53, 1.40; high CoE). Treatment with ivermectin may reduce progression to severe disease; however, the evidence failed to demonstrate a beneficial or detrimental effect on symptoms (RR: 0.70; 95% CI: 0.44, 1.11; moderate CoE). Treatment with ivermectin failed to demonstrate a beneficial or detrimental effect on hospitalization or viral clearance at day seven (RR: 0.88; 95% CI: 0.71, 1.11, moderate CoE, and RR: 1.01; 95% CI: 0.78, 1.31; very low CoE, respectively). The evidence is very uncertain due to the inclusion of one study without appropriate randomization, but ivermectin may reduce the time to recovery among ambulatory persons with COVID-19 (mean difference: 2.99 days fewer; 95% CI: 4.76 to 1.22 days fewer; very low CoE). However, the ACTIV-6 trial did not show a reduction in time to recovery with a hazard ratio: 1.09 (0.98, 1.22) [212].
Harms
In doses typically used for the treatment of parasitic infections, ivermectin is well tolerated. We are unable to exclude the potential for serious adverse events in hospitalized patients and ambulatory persons with COVID-19 treated with ivermectin rather than no ivermectin, (RR: 1.03; 95% CI: 0.32, 3.34; moderate CoE and RR: 0.81; 95% CI: 0.51, 1.30; moderate CoE, respectively).
Other considerations
The panel determined the certainty of evidence of treatment of ivermectin for hospitalized patients to be very low due to concerns with risk of bias (i.e., study limitations) and imprecision. However, the panel’s decision for hospitalized patients was indirectly informed by the lack of benefit of ivermectin as seen in studies in ambulatory persons. The panel determined the certainty of evidence of treatment of ivermectin for ambulatory persons to be moderate due to concerns with imprecision. The guideline panel made a conditional recommendation against treatment of COVID-19 with ivermectin outside of the context of a clinical trial for both patients with COVID-19 hospitalized or in the outpatient setting.
Conclusions and research needs for this recommendation
The guideline panel suggests against ivermectin for the treatment of hospitalized patients with COVID-19. The guideline panel recommends against ivermectin for the treatment of outpatients with COVID-19.
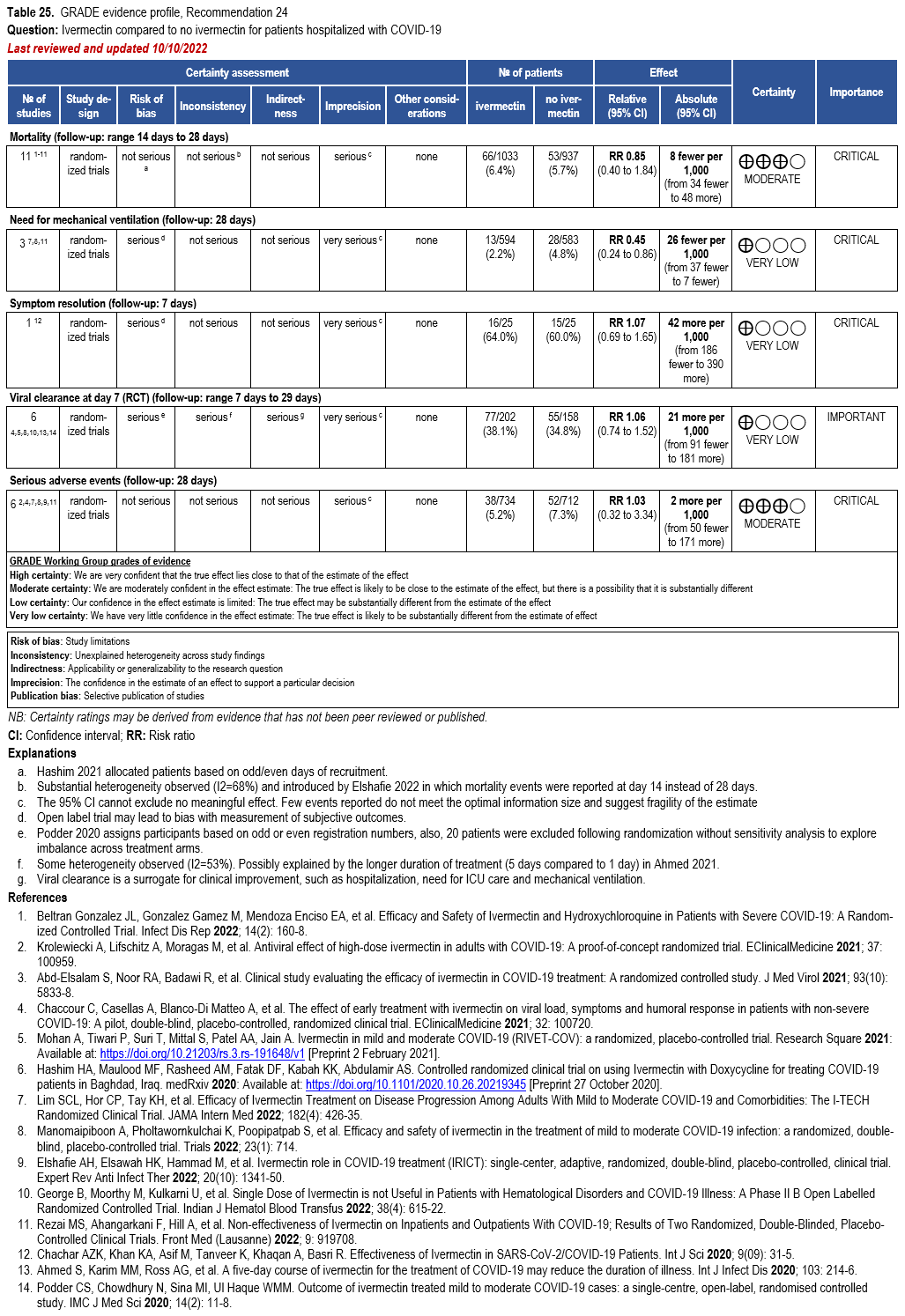
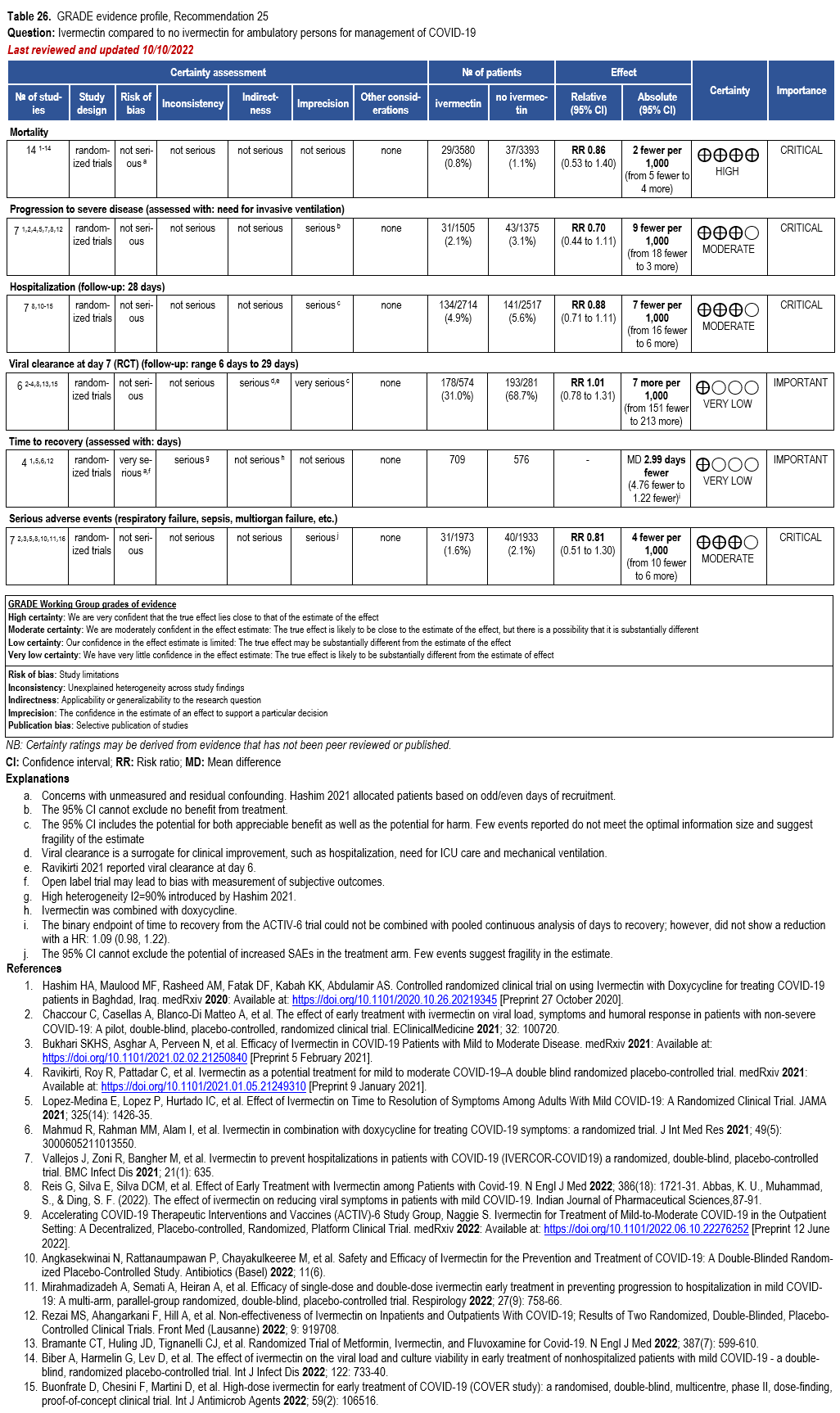
Supplementary Materials
Study characteristics
Forest Plots
- Figure s9a. Mortality for ivermectin vs. no ivermectin among hospitalized patients (from RCTs)?
- Figure s9b. Need for mechanical ventilation for ivermectin vs. no ivermectin among hospitalized patients
- Figure s9c. Viral clearance at seven days for ivermectin vs. no ivermectin among hospitalized patients (all studies)
- Figure s9d. Viral clearance at seven days for ivermectin vs. no ivermectin among hospitalized patients (without Ahmed 2020)
- Figure s9e. Serious adverse events for ivermectin vs. no ivermectin among hospitalized patients
- Figure s9f. Mortality for ivermectin vs. no ivermectin among ambulatory patients
- Figure s9g. Progression to severe disease for ivermectin vs. no ivermectin among ambulatory patients
- Figure s9h. Viral clearance at seven days for ivermectin vs. no ivermectin among ambulatory patients
- Figure s9i. Time to recovery for ivermectin vs. no ivermectin among ambulatory patients
- Figure s9j. Hospitalization for ivermectin vs. no ivermectin among ambulatory patients
- Figure s9k. Serious adverse events for ivermectin vs. no ivermectin among ambulatory patients
Risk of bias
Recommendation 26: Fluvoxamine
Section last reviewed and updated 11/8/2021
Last literature search conducted 10/31/2021
Recommendation 26: Among ambulatory patients with COVID-19, the IDSA guideline panel recommends fluvoxamine only in the context of a clinical trial. (Knowledge gap)
Why is fluvoxamine considered for treatment?
Fluvoxamine is a selective serotonin reuptake inhibitor (SSRI) which is currently FDA-approved for the treatment of obsessive-compulsive disorder. SSRIs have been shown to have affinity for Sigma-1 receptors, which have been demonstrated to modulate cytokine levels in animal models of septic shock [228]. Additionally, pharmacologic agents that act at Sigma-1 receptors have demonstrated in vitro activity against SARS-CoV-2 [229]. Amongst the SSRIs, fluvoxamine has been shown to have the high affinity for these receptors making it a potential repurposed drug option for the management of COVID-19 [230]. SSRIs like fluvoxamine may decrease uptake of serotonin from platelets during thrombosis, resulting in decreased neutrophil recruitment and platelet aggregation, which may be helpful in the early stages of COVID-19 [231, 232].
Summary of the evidence
Our search identified two RCTs that reported on ambulatory patients with SARS-CoV-2 infection [233, 234]. Patients in these studies were randomized to fluvoxamine or placebo/usual care. Both trials included symptomatic outpatients who tested positive for SARS-CoV-2 infection within seven days. Reis included patients who were at high risk for severe infection and utilized a composite primary outcome of hospitalization or emergency room visit lasting greater than six hours [234]. Additional outcomes reported in the two trials included mortality, hospitalization, emergency room visit lasting >6 hours, progression to oxygen saturation <92%, viral clearance, and serious adverse events.
Benefits
Outpatients
Among symptomatic ambulatory patients with COVID-19, fluvoxamine failed to demonstrate or to exclude a beneficial effect on mortality at 28 days compared to no fluvoxamine (RR: 0.69; 95% CI: 0.38, 1.27; low CoE). Fluvoxamine showed a reduction of the composite outcome of hospitalizations, emergency room visits lasting >6 hours, or oxygen saturation <92% (RR: 0.64; 0.50, 0.84; low CoE). When evaluating the effect on hospitalizations only, there was a trend toward less hospitalizations in fluvoxamine treated patients compared to those not receiving fluvoxamine (RR: 0.75; 95% CI: 0.57, 0.99; low CoE). Treatment with fluvoxamine failed to show a benefit in viral clearance at day seven (RR: 0.74; 0.52, 1.05; very low CoE).
Harms
The risk of serious adverse events in patients receiving fluvoxamine was not greater than those not receiving fluvoxamine (RR: 0.81; 95% CI: 0.59, 1.12; low CoE).
Other considerations
The panel agreed on the overall low certainty of evidence given the sparseness in mortality data and because upper boundary of the 95% confidence interval failed to exclude the risk of possible harms. The panel also had concerns about the generalizability/indirectness in the results surrounding hospitalization and emergency room visit >6 hours as one study [234] was partially conducted in patients with extended stays in emergency settings (mobile hospitals) to inform the primary endpoint, and it is unclear if resource constraints (possible contingency setting) may have affected the total number of events (i.e., emergency room stays and rates of hospitalization).
Conclusions and research needs for this recommendation
The guideline panel recommends fluvoxamine only in the context of a clinical trial to better delineate the effects of fluvoxamine on disease progression, such as need for hospital admission, ICU care, and ultimately, mortality.
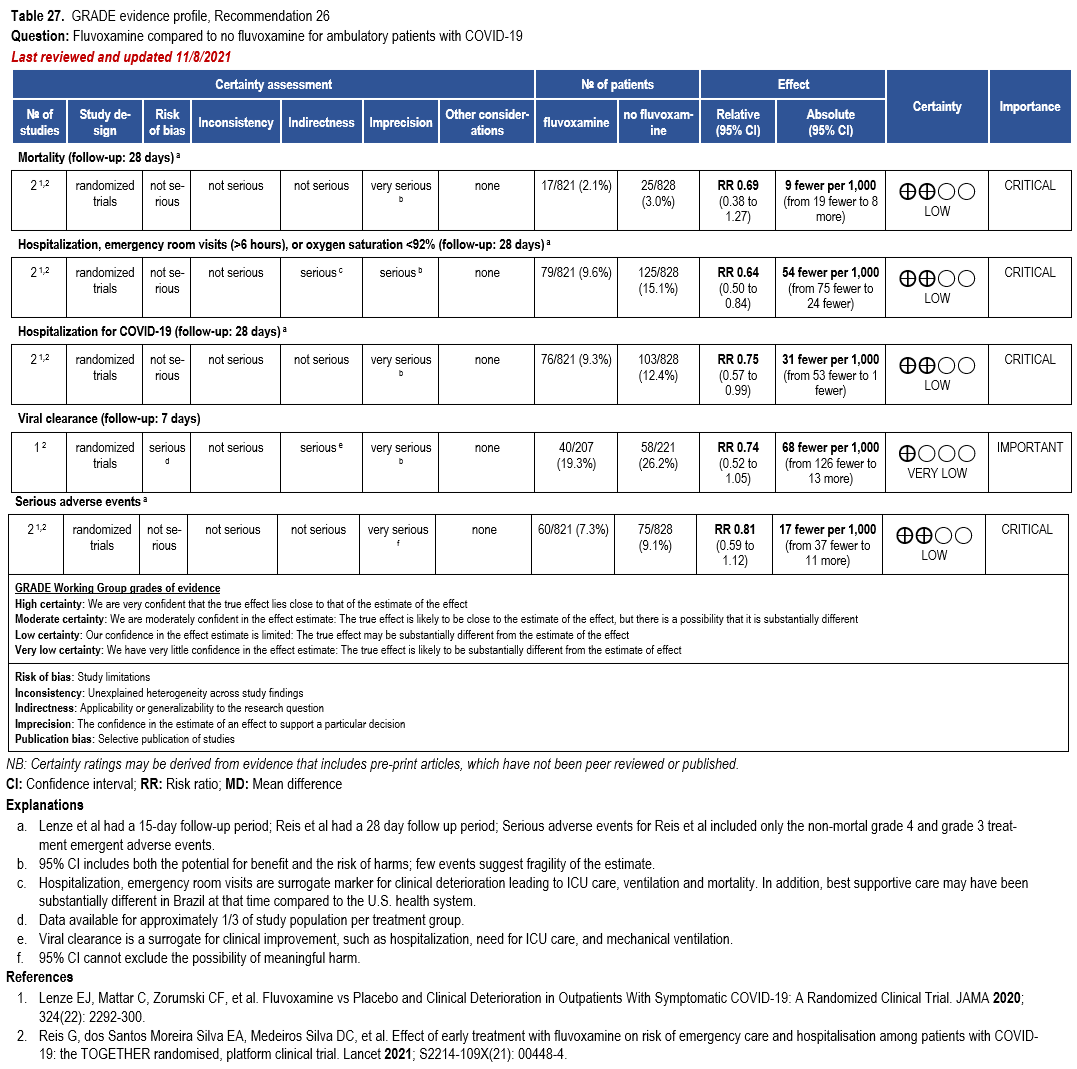
Supplementary Information
Study characteristics:
Forest plots:
- Figure s10a. Outcome of mortality for fluvoxamine vs. no fluvoxamine
- Figure s10b. Outcomes of hospitalization, emergency room visits (>6 hours), or oxygen saturation <92% for fluvoxamine vs. no fluvoxamine
- Figure s10c. Outcome of hospitalization for fluvoxamine vs. no fluvoxamine
- Figure s10d. Outcome of serious adverse events for fluvoxamine vs. no fluvoxamine
Risk of bias:
Recommendation 27: Nirmatrelvir/ritonavir
Section last reviewed and updated 4/12/2023
Last literature search conducted 3/31/2023
Resources:
- University of Liverpool: COVID-19 drug interaction checker
- University of Liverpool: HIV drug interaction checker
Recommendation 27(UPDATED 4/12/2023): In ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease, the IDSA guideline panel suggests nirmatrelvir/ritonavir initiated within five days of symptom onset rather than no nirmatrelvir/ritonavir. (Conditional recommendation†, Low certainty of evidence)
Remarks:
- Patients’ medications need to be screened for serious drug interactions
- Dosing based on renal function:
- Estimated glomerular filtration rate (eGFR) > 60 ml/min: 300 mg nirmatrelvir/100 ritonavir every 12 hours for five days
- eGFR ≤60 mL/min and ≥30 mL/min: 150 mg nirmatrelvir/100 mg ritonavir every 12 hours for five days
- eGFR <30 mL/min: not recommended
- Patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease admitted to the hospital may also receive nirmatrelvir/ritonavir
*Options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, remdesivir for a 3-day course, molnupiravir, and neutralizing monoclonal antibodies. Patient-specific factors (e.g., symptom duration, renal function, drug interactions) as well as product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.

Why is nirmatrelvir/ritonavir considered for treatment?
Nirmatrelvir is an inhibitor to the main protease (Mpro) of SARS-CoV-2; inhibition of this enzyme blocks viral replication. Nirmatrelvir is a substrate of the cytochrome P450 3A4 isoenzyme system and is co-packaged with an HIV-1 protease inhibitor, ritonavir, a potent inhibitor of cytochrome P450 3A4. Coadministration results in higher concentrations and a longer half-life of nirmatrelvir, allowing for every-12-hour dosing. The FDA granted EUA to nirmatrelvir/ritonavir on December 22, 2021 for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (≥12 years of age and weighing ≥40 kg) who are at high risk for progression to severe COVID-19, including hospitalization or death [235].
Summary of the evidence
Our search identified one RCT reporting on treatment of mild-to-moderate COVID-19 in patients at high risk for progression to severe disease [235]. In addition, the search identified one RCT reporting on treatment of mild-to-moderate COVID-19 in 264 hospitalized patients [236]. Some data used to prepare this recommendation were extracted from the FDA EUA document.
Benefits
Nirmatrelvir/ritonavir
All-cause mortality through day 28 may be lower in ambulatory patients receiving nirmatrelvir/ritonavir compared to no nirmatrelvir/ritonavir (RR: 0.04; 95% CI: 0.00, 0.69; low CoE). Patients treated with nirmatrelvir/ritonavir rather than no nirmatrelvir/ritonavir may have fewer COVID-19-related hospitalizations (RR: 0.12; 95% CI: 0.06, 0.26; low CoE). The composite endpoint of COVID-19-related hospitalizations or mortality was lower in patients receiving nirmatrelvir/ritonavir compared to no nirmatrelvir/ritonavir (RR: 0.12; 95% CI: 0.06, 0.25; low CoE).
In hospitalized patients receiving nirmatrelvir/ritonavir, all-cause mortality may be lower (RR: 0.63; 95% CI: 0.21, 1.86; low CoE); however, no benefit has been shown for need for invasive mechanical ventilation or length of hospital stay (RR: 1.67; 95% CI: 0.62, 4.45; low CoE and MD -0.38 days; 95% CI: -2.09, 1.32; low CoE, respectively.
Harms
Nirmatrelvir/ritonavir
Limited evidence from hospitalized patients with mild-to-moderate COVID-19 receiving nirmatrelvir/ritonavir suggest increased serious adverse events and adverse events (RR 1.20; 95% CI: 0.38, 3.84; low CoE and RR: 1.40; 95% CI: 0.65, 3.04; low CoE).
Serious treatment-emergent adverse events were not reported in the FDA EUA.
Given co-formulation with ritonavir as a pharmacokinetic booster, there is potential for significant drug interactions. Contraindications exist between agents that can have their levels increased or decreased by nirmatrelvir and/or ritonavir and agents that can increase the metabolism of the components of nirmatrelvir and/or ritonavir, resulting in a loss of virologic response and possible resistance. These drug interactions can result in treatment failure or serious adverse events, which may lead to severe, life-threatening, or fatal events from greater exposures (i.e., higher levels) of concomitant medications. See Figures 2 and 3.


Less severe but clinically meaningful drug interactions may also occur when nirmatrelvir/ritonavir is co-administered with other agents. Levels of immunosuppressive agents such as tacrolimus, cyclosporine, or sirolimus can be increased when administered with nirmatrelvir/ritonavir. Hormonal contraceptives containing ethinyl estradiol may possibly have reduced effectiveness due to lowered ethinyl estradiol levels when administered with nirmatrelvir/ritonavir. Women of childbearing potential should be counseled to use a back-up, non-hormonal method of contraception.
Patients with moderate renal impairment (eGFR <60 and ≥30 mL/min) must be counseled that they will only take one 150-mg nirmatrelvir tablet (oval shape, pink) with one 100-mg tablet of ritonavir twice daily, instead of the regular dose of two 150-mg nirmatrelvir (300 mg) tablets with one 100-mg tablet of ritonavir twice daily. Pharmacists need to adhere to the specific instructions when dispensing the product according to instructions provided in the EUA [237]. Given the lack of renal function/eGFR data at the point of dispensing, providers must specify the numeric dosage of each agent on the prescription to ensure the correct dose is provided to the patient at the point of dispensing. There are no data in patients with severe renal disease (eGFR ≤ 30 mL/min); this medication is currently not recommended in patients with severe renal disease until more data on dosing in this population are available.
There are no dose adjustments needed for patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment, however, data are lacking in patients with Child-Pugh C, and nirmatrelvir/ritonavir is therefore not recommended in this population.
According to the EUA, nirmatrelvir/ritonavir use may be associated with a risk of developing HIV resistance to HIV protease inhibitors in individuals with uncontrolled or undiagnosed HIV-1 infection.
Other considerations
Nirmatrelvir/ritonavir
The panel agreed that the overall certainty of the evidence for the treatment of ambulatory patients was low; there are concerns with the inability to exclude potential risks to bias because of limited availability of study details within the EUA, and there is imprecision due to a low number of events reported. The EUA did not report safety data (e.g., adverse events or severe adverse events) from the trial. The panel agreed that the benefits are likely to outweigh any potential harms in patients with COVID-19 who are at high risk of severe disease; however, recognized concerns with drug interactions must be considered.
The evidence confirms that using nirmatrelvir/ritonavir early in the disease process when viral loads are high confers maximum benefit. It is critical to make a rapid diagnosis and treat ambulatory patients with COVID-19 early in the disease course. Observational studies have shown a similar benefit among vaccinated patients infected with newer variants. The panel recognized the need for additional evidence to inform decisions regarding treatment of hospitalized patients with COVID-19.
Viral rebound in patients treated with nirmatrelvir/ritonavir
Recurrence of symptoms associated with viral rebound has been estimated to occur in nirmatrelvir/ritonavir treated patients in 0.8% to 6.6% in various trials, including the EPIC HR trial [238, 239]. Rebound has also been described with molnupiravir (5.8% [240] and no antiviral treatment [238, 241]). Observational evidence suggests hospitalization after nirmatrelvir/ritonavir treatment to be infrequent, ranging from 0.11% to 0.44% [242, 243]. No direct evidence was found on the effect of repeat nirmatrelvir/ritonavir treatment (on any other direct acting antivirals) in patients experiencing symptomatic viral rebound after initial antiviral treatment. The effect of repeating the same drug (for another course) after a viral rebound is unknown regards to patient important outcomes such as need for hospitalization, invasive ventilation, or death. Study limitations of observational medical records database studies includes misclassifications in admission diagnosis and absence of adequate compliance determination, among others.
Conclusions and research needs for this recommendation
Nirmatrelvir/ritonavir
The guideline panel suggests the use of nirmatrelvir/ritonavir for ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who are within five days of symptom onset. More data are needed on the potential adverse effects of this medication. In addition, future studies are important to inform the impact of nirmatrelvir/ritonavir in hospitalized patients, in vaccinated high-risk patients with mild-to-moderate COVID-19 and in symptomatic immune- compromised patients with persistently elevated viral loads.
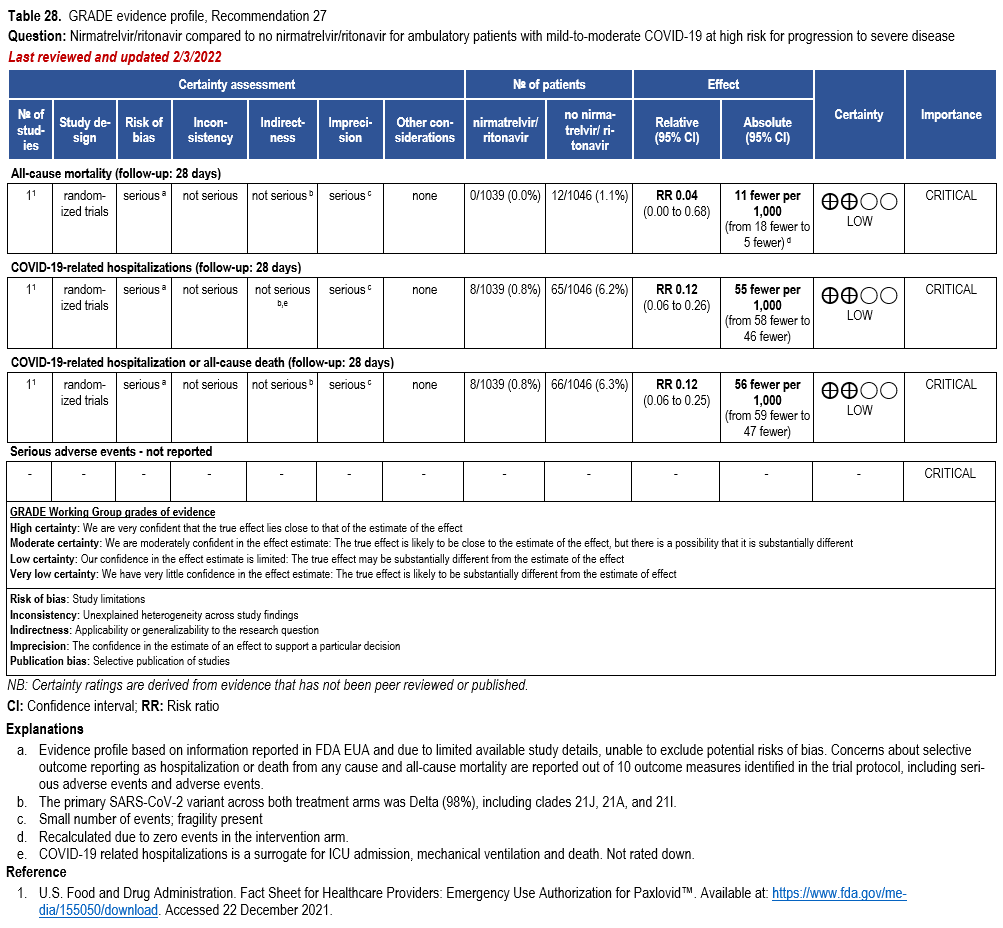
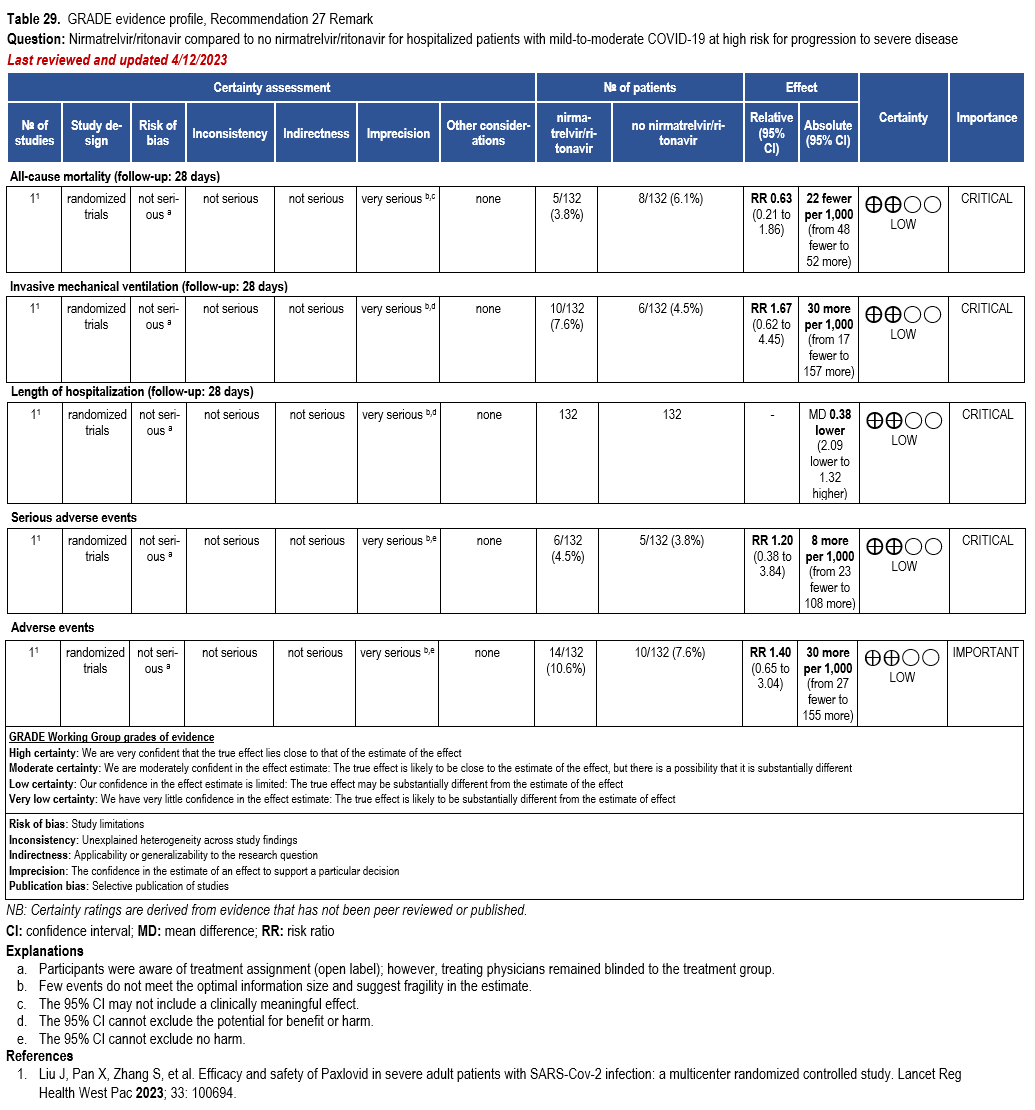
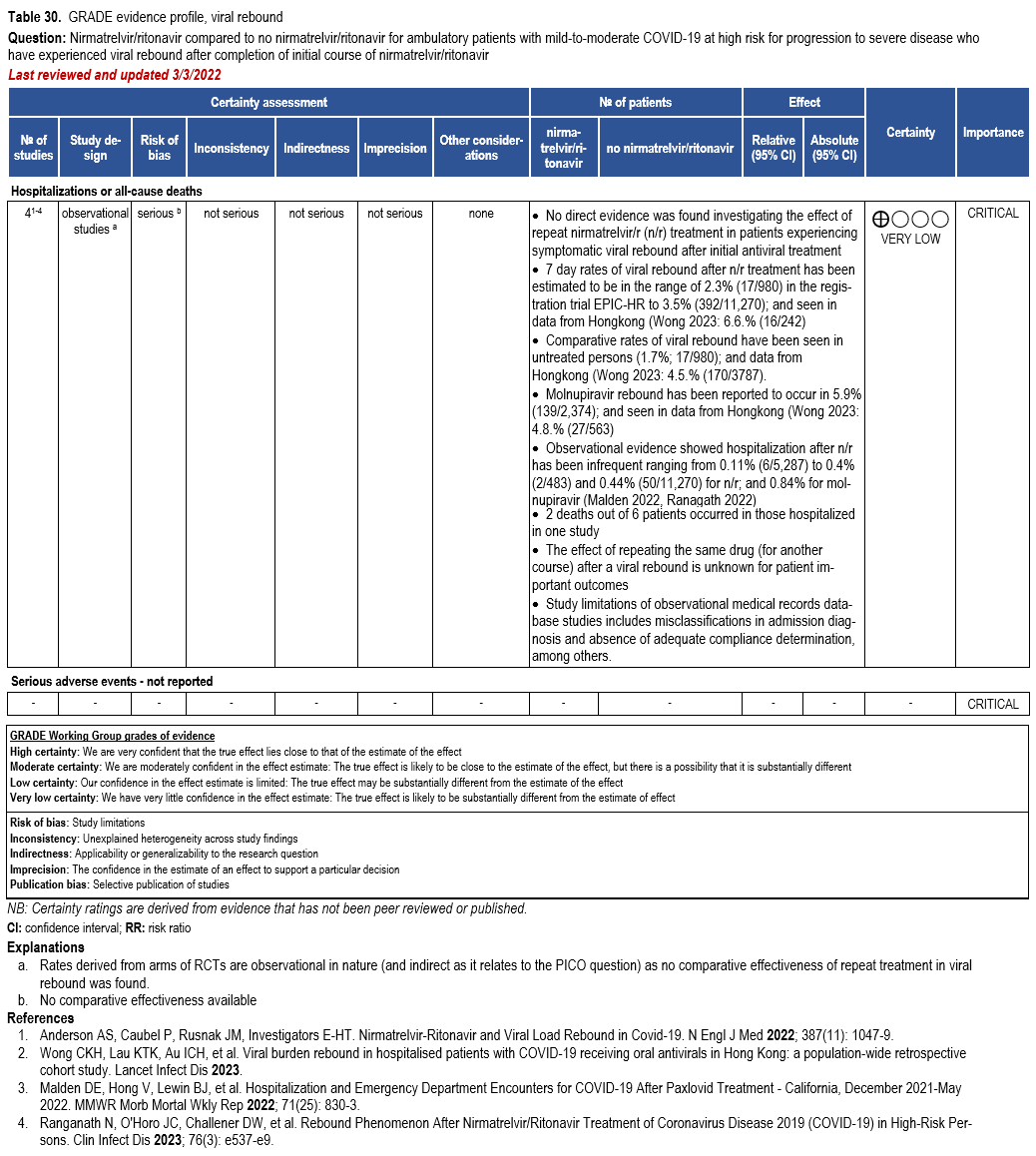
Supplementary Information
Study characteristics:
Risk of bias:
Recommendation 28: Molnupiravir
Section last reviewed and updated 2/23/2023
Last literature search conducted 1/31/2023
Recommendation 28: In ambulatory patients (≥18 years) with mild-to-moderate COVID-19 at high risk for progression to severe disease who have no other treatment options*, the IDSA guideline panel suggests molnupiravir initiated within five days of symptom onset rather than no molnupiravir. (Conditional recommendation†, Low certainty of evidence)
*Other options for treatment and management of ambulatory patients include nirmatrelvir/ritonavir, three-day treatment with remdesivir, Patient-specific factors (e.g., symptom duration, renal function, drug interactions) as well as product availability should drive decision-making regarding choice of agent. Data for combination treatment do not exist in this setting.
Remarks:
- Patients who will most likely benefit from antivirals are those with risk factors for progression to severe disease (e.g., elderly, those with high-risk comorbidities, incomplete vaccination status, or immunocompromised). Those without risk factors are less likely to benefit.
- Patients who put a higher value on the putative mutagenesis, adverse events, or reproductive concerns and a lower value on the uncertain benefits would reasonably decline molnupiravir.
- Patients with mild-to-moderate COVID-19 who are at high risk of progression to severe disease admitted to the hospital for reasons other than COVID-19 may also receive molnupiravir.
- Molnupiravir is not authorized under the FDA EUA for use in patients <18 years because it may affect bone and cartilage growth.
- Molnupiravir is not recommended under the FDA EUA for use during pregnancy.
- Molnupiravir is not authorized under the FDA EUA for pre-exposure or post-exposure prevention of COVID-19 or for initiation of treatment in patients hospitalized due to COVID-19 because benefit of treatment has not been observed in individuals when treatment is started after hospitalization due to COVID-19.
†The guideline panel concluded that the desirable effects outweigh the undesirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.

Why is molnupiravir considered for treatment?
Molnupiravir is an oral antiviral that targets the genetic machinery that is responsible for SARS COV-2 replication. Molnupiravir is an oral pro-drug that is converted to β-D-N4-hydroxycytidine, which acts as a substrate for RNA-dependent RNA polymerase. After it is incorporated into the viral RNA, serial mutations develop, resulting in a virus that is less fit for ongoing viral replication. One phase I RCT evaluated the safety and tolerability of molnupiravir in healthy adults without COVID-19 [235]. The study reported molnupiravir to be well tolerated, with no increased reports of serious adverse events among persons in the molnupiravir arm compared to those receiving placebo. The FDA granted EUA to molnupiravir on December 23, 2021, for the treatment of mild-to-moderate COVID-19 in adults (≥18 years) who are at high risk for progression to severe COVID-19, including hospitalization or death.
Summary of the evidence
Five RCTs informed the recommendation for molnupiravir [245-249]. Three RCTs re-ported on treatment of at least partially vaccinated participants with COVID-19 with either 800 mg of molnupiravir or placebo on outcomes of mortality, hospitalization, and serious adverse events [246, 247, 249]. In the largest trial (N=26,411), PAMORAMIC, 99% of participants had at least one COVID-19 vaccine dose with 92%-93% having received three doses [246]. Two RCTs reported on treatment of unvaccinated patients with COVID-19 with either 800 mg of mol-nupiravir or placebo for five days [245, 248]. In one phase III trial (MOVe-OUT trial) reporting on the outcomes of death, hospitalization and serious adverse events, patients with mild-to-moderate COVID-19 received either molnupiravir or placebo within five days after the onset of symptoms. In the phase IIa trial reporting on the outcomes of death and serious adverse events in patients with symptom duration <7 days received molnupiravir or placebo.
Benefits
COVID-19-related mortality may be lower in patients receiving molnupiravir rather than placebo (RR: 0.28; 95% CI: 0.09, 0.86; low CoE); however, given the small baseline risk of mortality across the available evidence, the reduction in mortality may not be clinically meaningful (Absolute effect: 1 fewer per 1,000 persons; 95% CI: from 1 fewer to 0 fewer). COVID-19-related hospitalizations and the composite of all-cause hospitalization or death likely results in little to no difference among patients receiving molnupiravir rather than no molnupiravir (RR: 1.03; 95% CI: 0.78, 1.35; moderate CoE and RR: 0.92; 95% CI: 0.74, 1. 14; moderate CoE, respectively).
Harms
Patients treated with molnupiravir may not experience greater serious adverse events or adverse events than those receiving placebo (RR: 0.57; 95% CI: 0.22, 1.52; moderate CoE and RR: 0.81; 95% CI: 0.47, 1.40; moderate CoE, respectively).
Based on findings from animal reproduction studies, molnupiravir may cause fetal harm when administered to pregnant individuals [250]. Other concerns with molnupiravir include the possibility of viral mutagenesis in persons with compromised immune systems who are unable to clear the virus. Females of childbearing potential should be counseled to use a reliable method of contraception during treatment and for four days after the last dose. Breastfeeding is not recommended during treatment with molnupiravir. Lactating individuals may consider interrupting breastfeeding and may consider pumping and discarding breast milk during treat-ment and for four days after last dose of molnupiravir [251]. Men of reproductive potential who are sexually active with females of childbearing potential should be counseled to use a reliable method of contraception during treatment and for at least three months after the last dose of molnupiravir. It is also not recommended in children <18 years of age for the concern of bone growth.
Molnupiravir does not require renal or hepatic dose adjustment.
Other considerations
The panel agreed that the overall certainty of evidence for treatment of ambulatory patients was low, given concerns with imprecision, driven by few reported events and a relatively small effect.
The use of molnupiravir presents additional considerations and potential concerns regarding viral mutagenesis in immunocompromised persons and safety in persons of reproductive age, for which more data are needed to quantify such effects. The panel recognized that alternative treatment options exist with the possibility of greater benefit with a smaller known safety profile. The FDA required the manufacturers to conduct additional animal studies on the impact of the drug on spermatogenesis and to establish a pregnancy registry if the drug was inadvertently administered during pregnancy.
The evidence confirms that using molnupiravir early in the disease process when viral loads are high confers maximum benefit. It is critical to make a rapid diagnosis and treat ambulatory patients with COVID-19 early in the disease course.
More recent studies in mild-to-moderate COVID-19 have shown lower rates of progression to hospitalizations or death, which could likely be due to changes in population immunity and lower virulence of recent circulating variants. Given this observation, the panel discussed about the role of patient centered outcomes (e.g., meaningful decrease in severity or duration of symptoms) other than mortality and hospitalizations in trials evaluating treatment of mild to moderate COVID-19. The panel agreed that such outcomes should be evaluated in double-blind placebo-controlled trials to reduce the risk of bias. Such outcomes also should be measured using validated instruments and should be coupled with measures of disability or quality of life. The studies evaluating molnupiravir which reported such outcomes had a high risk of bias so were not considered for making the recommendation.
Conclusions
The guideline panel suggests the use of molnupiravir for ambulatory patients with mild-to-moderate COVID-19 at high risk for progression to severe disease who are within five days of symptom onset and have no other treatment options. More data are needed on the potential adverse effects of this medication. The evidence supporting this recommendation will be reassessed with the release of updated published information from newer trials.
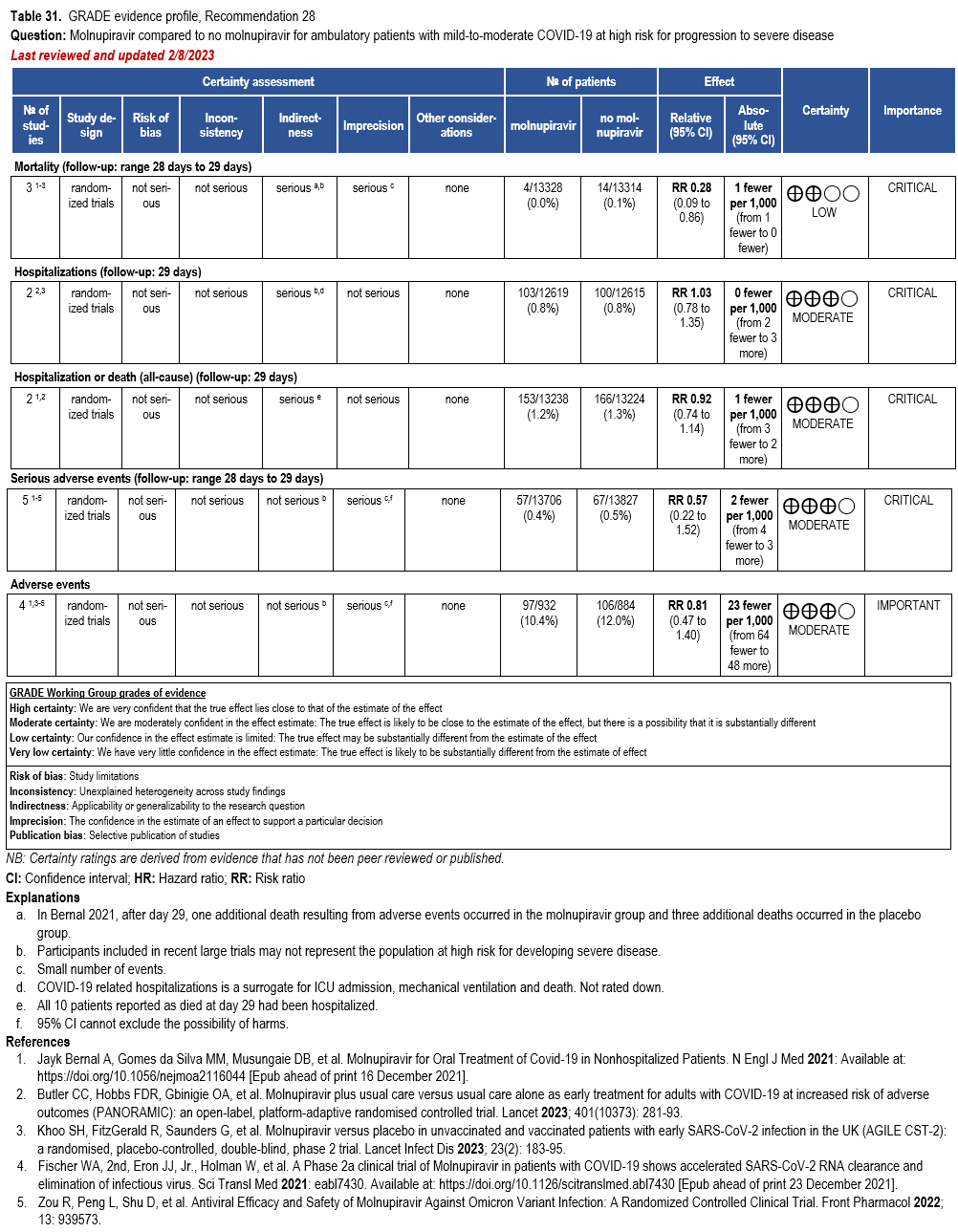
Supplementary Information
Study characteristics:
Forest plots:
- Figure s11a. Forest plot for the outcome of mortality for molnupiravir vs. no molnupiravir
- Figure s11b. Forest plot for the outcome of hospitalization for molnupiravir vs. no molnupiravir
- Figure s11c. Forest plot for the outcome of hospitalization or death for molnupiravir vs. no molnupiravir
- Figure s11d. Forest plot for the outcome of serious adverse events for molnupiravir vs. no molnupiravir
- Figure s11e. Forest plot for the outcome of adverse events for molnupiravir vs. no molnupiravir
Risk of bias:
Recommendations 29-30: Colchicine
Section last reviewed and updated 6/30/2022
Last literature search conducted 5/31/2022
Recommendation 29: In hospitalized patients with COVID-19, the IDSA panel recommends against colchicine for treatment of COVID-19. (Strong recommendation, Moderate certainty of evidence)
Recommendation 30: In ambulatory persons with COVID-19, the IDSA panel suggests against colchicine for treatment of COVID-19. (Conditional recommendation††, Moderate certainty of evidence)
††The guideline panel concluded that the undesirable effects outweigh the desirable effects, though uncertainty still exists, and most informed people would choose the suggested course of action, while a substantial number would not.
Why is colchicine considered for treatment?
Colchicine has been used in various inflammatory conditions, such as gouty arthritis, pericarditis, and familial Mediterranean fever for its anti-inflammatory properties. The anti-inflammatory mechanisms of colchicine are broad [252, 253] and include disruption of micro-tubules resulting in downregulation of pro-inflammatory cytokines [254, 255] and by reducing recruitment of inflammatory cells to endothelial cells [256]. Colchicine is widely available and relatively cheap, making it an attractive therapeutic to mitigate the inflammatory phase of COVID-19. This has resulted in numerous randomized controlled trials of colchicine in the man-agement of COVID-19.
Summary of the evidence
Our search identified 12 comparative randomized controlled trials in persons with COVID-19 treated with colchicine or an inactive comparison (e.g., standard of care with or without place-bo). Ten studies [257-266] informed the recommendations for hospitalized patients and report-ed on the outcomes of mortality, need for mechanical ventilation, length of hospital stay, and adverse events. The three studies [266-268] identified to inform the recommendation for am-bulatory persons reported on the outcomes of mortality, hospitalization, need for mechanical ventilation, and serious adverse events.
Benefits
Hospitalized
In hospitalized patients, treatment with colchicine for COVID-19 rather than no colchicine failed to show or exclude a beneficial effect on mortality (RR; 95% CI: 0.99; 0.92, 1.06; moderate CoE). Treatment with colchicine rather than no colchicine for the purpose of COVID-19 does not reduce need for mechanical ventilation (RR: 1.02; 95% CI: 0.90, 1.16; high CoE). Hospitalized patients receiving colchicine experienced a trend toward reduced hospital stay (MD: -1.77 days; 95% CI: -3.69, 0.15; very low CoE); however, there are concerns about risk of bias, inconsistency and imprecision.
Ambulatory
Treatment with colchicine likely does not reduce mortality or need for mechanical ventilation compared to no colchicine among ambulatory persons with COVID-19 (RR: 0.50; 95% CI: 0.19, 1.33; moderate CoE and RR: 0.50; 95% CI: 0.24, 1.07, moderate CoE, respectively). The evidence failed to demonstrate a beneficial or detrimental effect on symptoms in hospitalization (RR: 0.82; 95% CI: 0.64, 1.05; moderate CoE).
Harms
Hospitalized
We were unable to exclude the potential for adverse events in hospitalized patients receiving treatment with colchicine rather than no colchicine for COVID-19 (RR: 2.04; 95% CI: 1.07, 3.91; low CoE).
Ambulatory
One study reported on serious adverse events among persons treated with colchicine rather than no colchicine for COVID-19. Serious adverse events may be less frequent among ambulatory persons receiving treatment with colchicine rather than no colchicine; however, this may not be meaningfully different from those not receiving colchicine (RR: 0.78; 95% CI: 0.61, 1.00; moderate CoE).
Other considerations
The panel determined the certainty of the evidence of treatment of colchicine for hospitalized patients to be moderate due to imprecision. The guideline panel made a strong recommendation against treatment of COVID-19 with colchicine for hospitalized patients with COVID-19.
The panel determined the certainty of the evidence of treatment of colchicine for ambulatory persons to be moderate due to imprecision. The guideline panel made a conditional recommendation against treatment of COVID-19 with colchicine for ambulatory persons.
Conclusions and research needs for this recommendation
The guideline panel recommends against colchicine for the treatment of hospitalized patients with COVID-19. The guideline panel suggests against colchicine for the treatment of ambulatory persons with COVID-19.
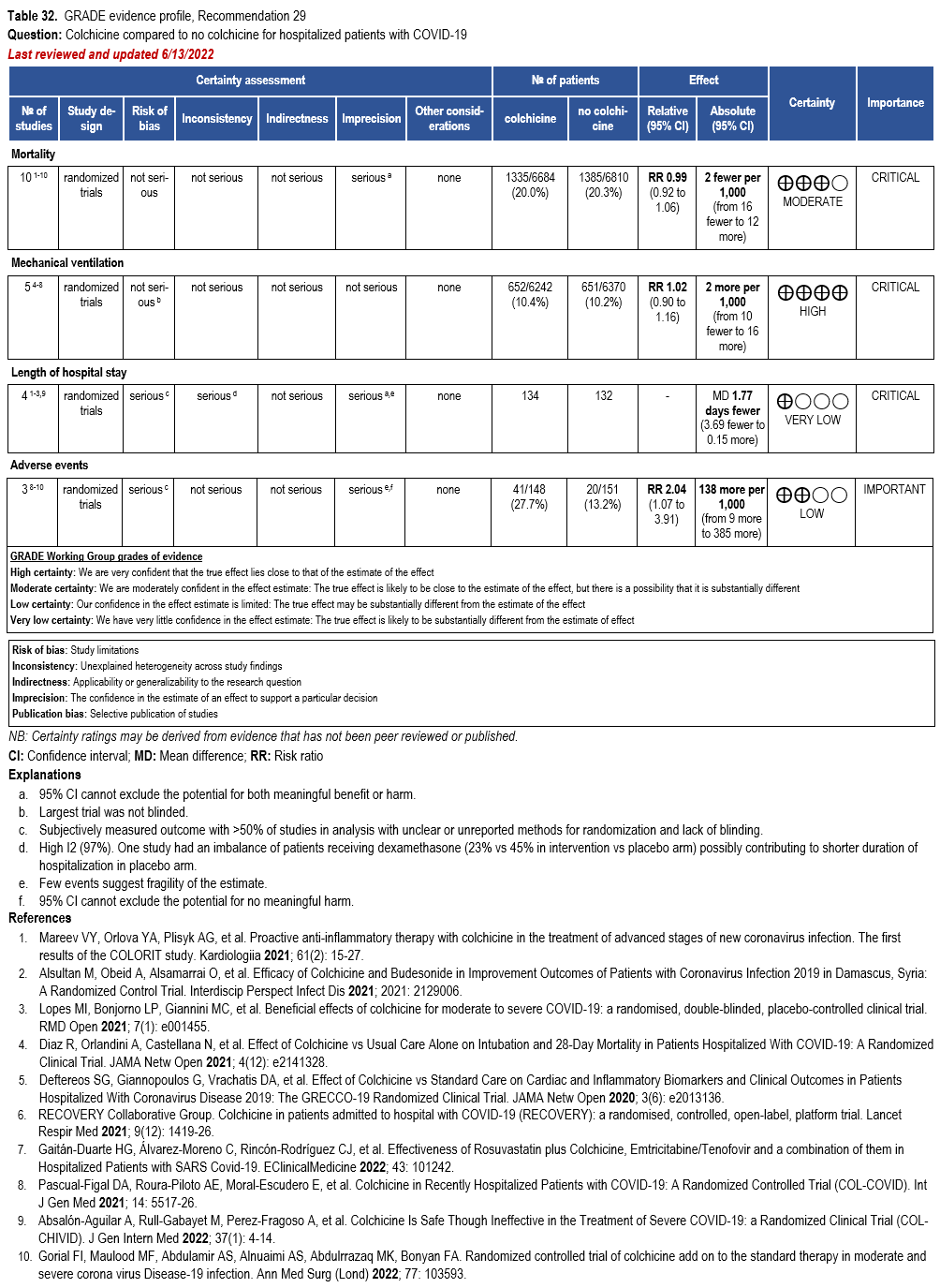
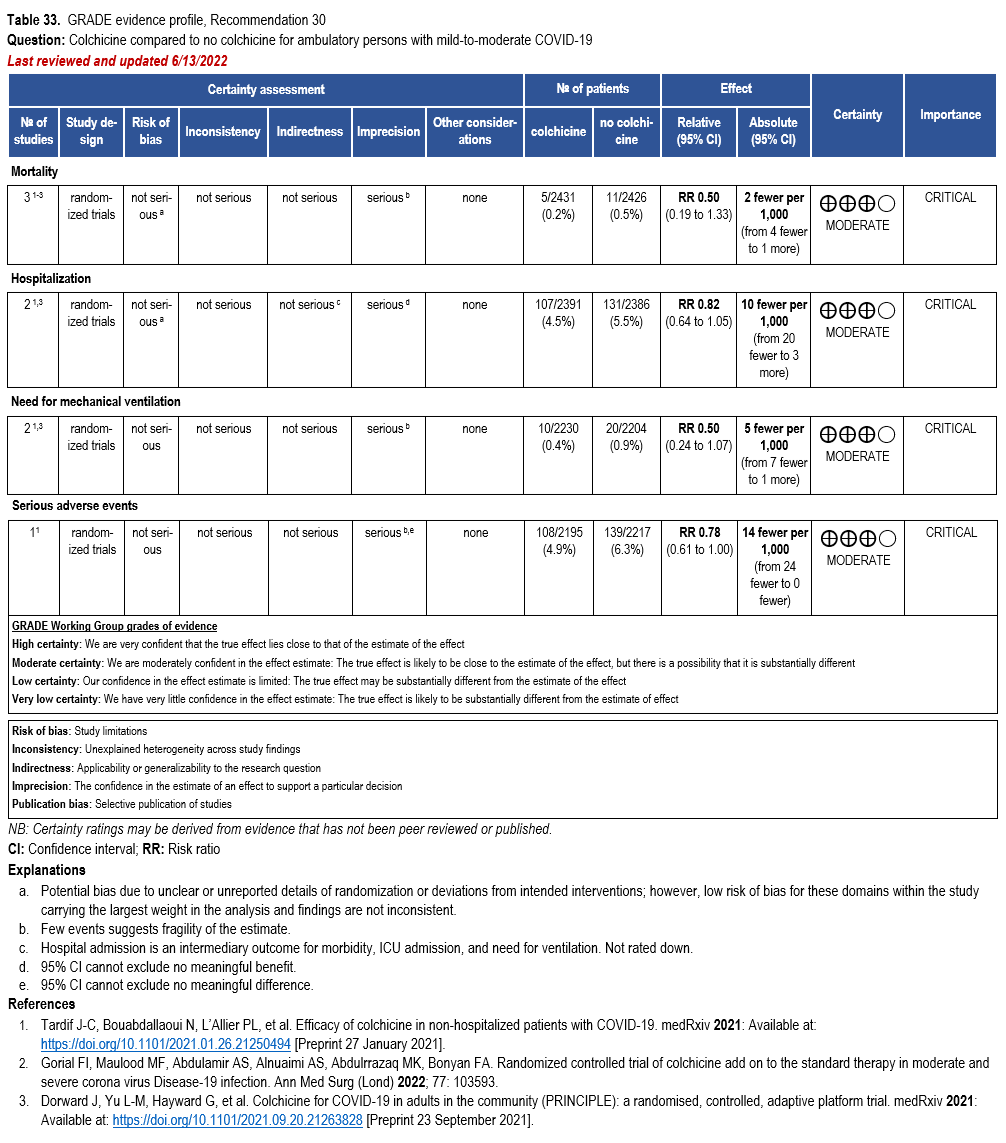
Supplementary Information
Study characteristics:
Forest plots:
- Figure s12a. Outcome of mortality for colchcine vs. no colchicine
- Figure s12b. Outcome of duration of hospitalization for colchicine vs. no colchicine (hospitalized patients)
- Figure s12c. Outcome of hospitalization for colchicine vs. no colchicine (ambulatory patients)
- Figure s12d. Outcome of mechanical ventilation for colchicine vs. no colchicine
- Figure s12e. Outcome of adverse events for colchicine vs. no colchicine (hospitalized patients)
Risk of bias:
Recommendation 31: Anakinra
Section last reviewed and updated 5/4/2023
Last literature search conducted 3/31/2023
Recommendation 31(NEW 5/4/2023): In hospitalized patients with severe* COVID-19, the IDSA guideline panel suggests against the routine use of anakinra. (Conditional recommendation, Low certainty of evidence)
Severity definitions:
*Severe illness is defined as patients with SpO2 ≤94% on room air, including patients on supplemental oxygen.
Why is anakinra considered for treatment?
Anakinra is a recombinant IL-1 inhibitor that is currently FDA approved for rheumatoid arthritis, cryopyrin-associated Periodic Syndromes, and Deficiency of IL-1 Receptor Antagonist. IL-1β is a pro-inflammatory cytokine, and in conjunction with other pro-inflammatory cytokines and interferon, can trigger hyperinflammation and ARDS which is often seen in severe COVID-19. Throughout the pandemic, Anakinra has been evaluated in numerous retrospective and randomized controlled trials for COVID-19. It was recently granted emergency use authorization by the US FDA for use in hospitalized adults with COVID-19 who require supplemental oxygen and who are at risk for progression to severe disease and are likely to have an elevated plasma suPAR (soluble urokinase plasminogen activator receptor) [269]. This authorization was based on results of the SAVE-MORE trial which utilized serum levels of the biomarker suPAR of ≥6 ng/mL to guide initiation of treatment with anakinra. The suPAR assay is not commercially available in the United States.
Summary of the evidence
Our search identified six RCTs that reported on hospitalized patients with severe COVID-19 who received treatment with either anakinra or no anakinra reporting on the outcomes of mortality, progression to mechanical ventilation, duration of hospital stay and serious adverse events [270-275].
Benefits
Hospitalized patients treated with anakinra showed a trend towards reduced progression to mechanical ventilation (RR: 0.69; 95% CI: 0.33, 1.44; low CoE). Anakinra appears to have trivial or no effect on mortality or duration of hospitalization (RR: 0.98; 95% CI: 0.57, 1.70; low CoE and MD: -0.93; 95% CI: -1.74, -0.11; low CoE, respectively).
Harms
Serious adverse events may not be meaningfully different among hospitalized patients treated with anakinra or not (RR: 0.93; 95% CI: 0.74, 1.19; low CoE).
Other considerations
The panel determined the certainty of evidence of treatment with anakinra for hospitalized patients with severe COVID-19 to be low due to concerns with imprecision, as effects failed to show or exclude a beneficial effect for mortality or duration of hospitalization. One study reported a reduction in mortality from treatment with anakinra used the suPAR scale to try to identify patients most likely to benefit from the treatment; however, this scale is not available in the US, restricting the feasibility of identifying the most appropriate patient group [272].
The guideline panel made a conditional recommendation against routine treatment with anakinra.
Conclusions and research needs for this recommendation
The guideline panel suggests against anakinra for the routine treatment of hospitalized patients with severe COVID-19. Research is needed to identify which patients are most likely to benefit from this drug in settings where suPAR testing is not available (e.g. the US). There are several therapies recommended for use in this population (link to Bari/Steroids?); however, this is a conditional recommendation against the use of anakinra based on low certainty of evidence, so in situations when other agents are not available patients who put a high value on the possible reduction in hospital stay and a low value on the uncertain effect on mortality would reasonably select treatment with anakinra.
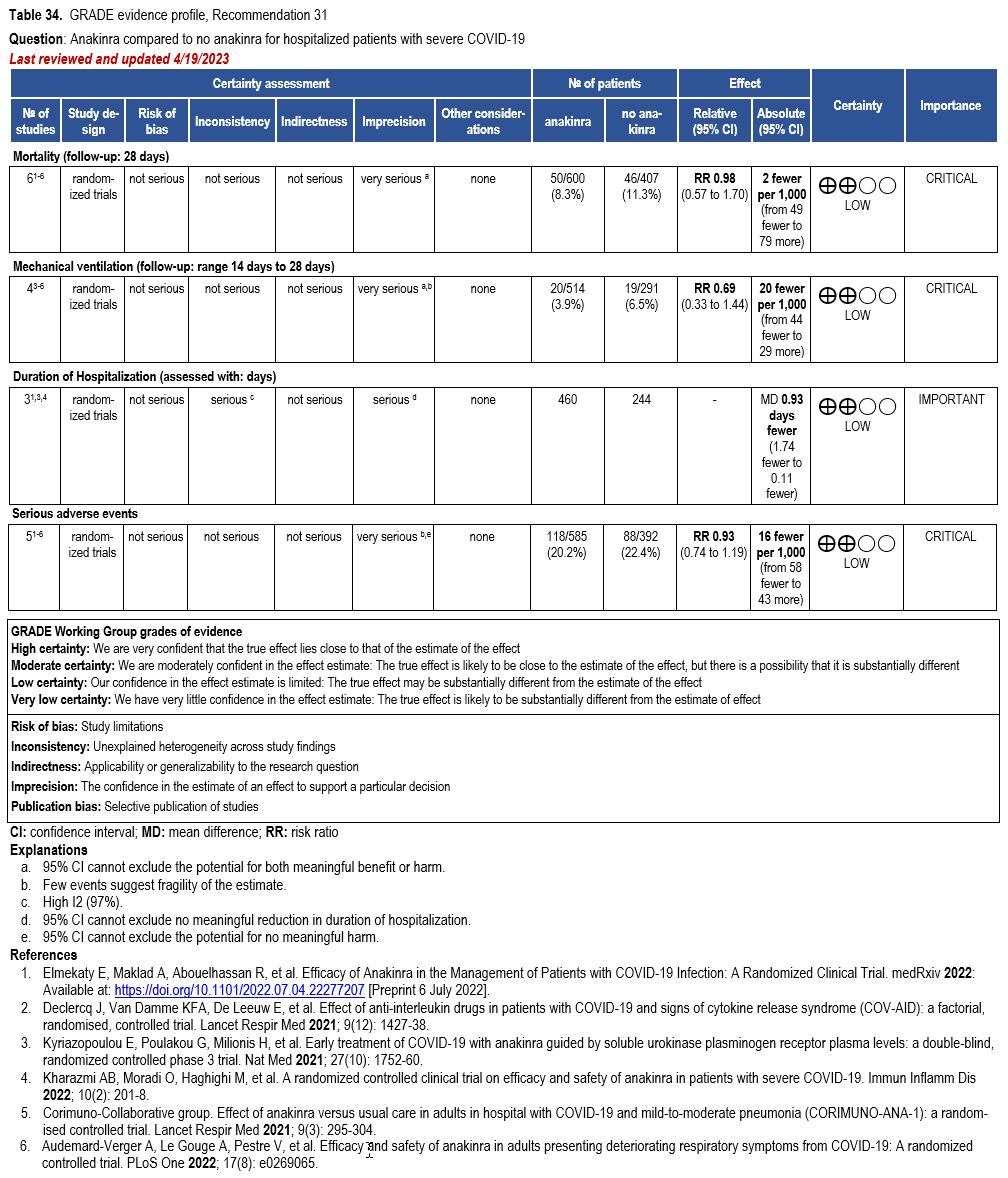
How to Approach a Patient When Considering Pharmacologic Treatments for COVID-19
In this section, we discuss how to approach a patient suspected to have COVID-19 and how to apply the IDSA COVID-19 treatment guidelines to specific clinical syndromes. The detailed evidence appraisals and recommendations for each therapeutic agent can be found in the individual sections. The certainty of supporting evidence is low to moderate for most recommendations; therefore, the guideline panel made conditional suggestions rather than strong recommendations for or against most of the agents. Though substantial progress was made with COVID-19 therapies in such a short period, there still remain many unanswered questions in the management of COVID-19. Therefore, the approach outlined here and in the guidelines are based on some assumptions and extrapolations. Despite limited evidence, to give actionable and timely guidance to frontline clinicians, we provide recommendations for use of combinations of agents, recommend some agents over others or extrapolate to sub populations not evaluated in trials.
Some of the critical unanswered questions in COVID-19 treatment trials are:
- Which sub-populations or specific clinical types of patients with COVID-19 benefit most from specific therapeutic agents?
- What is the efficacy and safety of COVID-19 therapies in populations that are immune from prior SARS-CoV-2 infections and vaccination?
- What is the efficacy and safety of treatments in infections with specific SARS-CoV-2 variants and sub-variants?
- How do therapeutic agents perform when compared to each other to allow a tiered approach to treating patients with COVID-19?
- What is the comparative efficacy and safety of nirmatrelvir/ritonavir versus remdesivir, molnupiravir, and different anti-SARS-CoV-2 antibodies in mild-to-moderate disease?
- What is the efficacy and safety of IL-6 inhibitors when compared to JAK inhibitors in severe disease?
- What is the comparative efficacy and safety of combinations of different drugs in treating different severities and clinical phenotypes of COVID-19?
- Which biomarkers can be used as predictors of therapeutic response to specific agents?
We hope future studies and trials address these uncertainties so we can give a more definitive treatment approach to COVID-19.
General principles of COVID-19 pharmacotherapy
During the early phase of the infection, when viral load is high and the host’s adaptive immune system has not mounted an adequate response, treatments targeting viral replication are most likely to be effective. These include both the direct antiviral therapies nirmatrelvir/ritonavir, molnupiravir, and remdesivir; and the passive immunity therapies of anti-SARS-CoV-2 antibodies and donor convalescent plasma. Timely initiation of antiviral therapies is critical as they are more efficacious when given within 5 to 7 days of symptom onset. Most patients do not progress to severe or critical disease, but some with risk factors do. Later in the disease process, in patients with severe and especially critical disease, an excessive and aberrant inflammatory response is implicated to be the primary cause of immunopathological damage. At this stage anti-inflammatory therapies like corticosteroids, IL-6 inhibitors or JAK inhibitors have been shown to be beneficial.
Clinical evaluation
Clinical evaluation should consider patient and pathogen specific factors that can influence choice of COVID-19 treatments. The evaluation should at least include assessment of:
- Severity of COVID-19
- Date of onset of symptoms
- Risk factors for progression to severe disease or death (see further discussion below, under Pharmacologic treatment of mild-to-moderate COVID-19 with risk factors for progression)
- Degree of chronic and acute end-organ dysfunction (including, but not limited to, pulmonary, cardiovascular, renal, and hepatic)
- Age and pregnancy status
- Virus-specific factors that may influence the choice of pharmacotherapy (e.g., variant specific susceptibility to certain drugs)
- Risk factors for progression are changing as the epidemic evolves with new variants, vaccination, and previous infection rates.
Diagnostic classification of severity of COVID-19 helps target specific treatments to patient populations that have been demonstrated to benefit in COVID-19 treatment trials. The clinician should identify which of the severity categories in Table 35 the patient falls into.
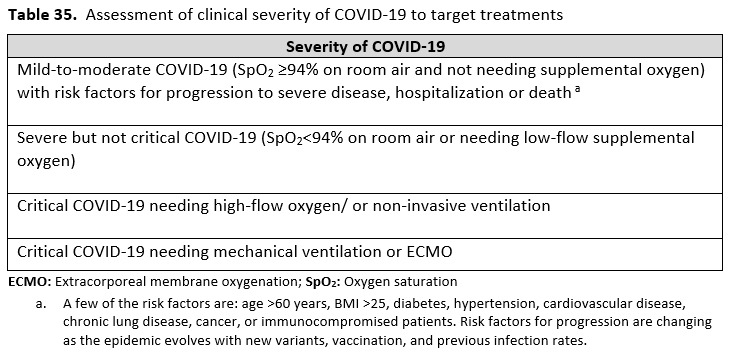
It is also important to identify factors that preclude the use of COVID-19 treatments or warrant their use with caution. Patients with low estimated GFR were not included in the trials for remdesivir and tocilizumab. Elevated aspartate transaminase (AST) and alanine transaminase (ALT) levels are a contraindication for IL-6 inhibitors and remdesivir. Patients who were neutropenic, had an active bacterial, fungal or parasitic infection, or were hypercoagulable were eliminated from some of the JAK inhibitor trials. It is also important to identify if the patients have other acute disease that either mimic COVID-19 or present concomitantly with COVID-19. Patients can have a positive SARS-CoV-2 by RT-PCR from a nasopharyngeal sample, and present with pulmonary disease caused by a bacterial pneumonia or pulmonary edema. Patients with COVID-19 can also have pulmonary embolism contributing to their symptoms and hypoxemia. It is important to avoid anchoring bias to the diagnosis of COVID-19 and be attentive to considering and evaluating other etiologies. Many of the COVID-19 therapies are not FDA-approved and have instead received FDA EUA, so it is necessary to follow the regulatory processes and protocols for these agents.
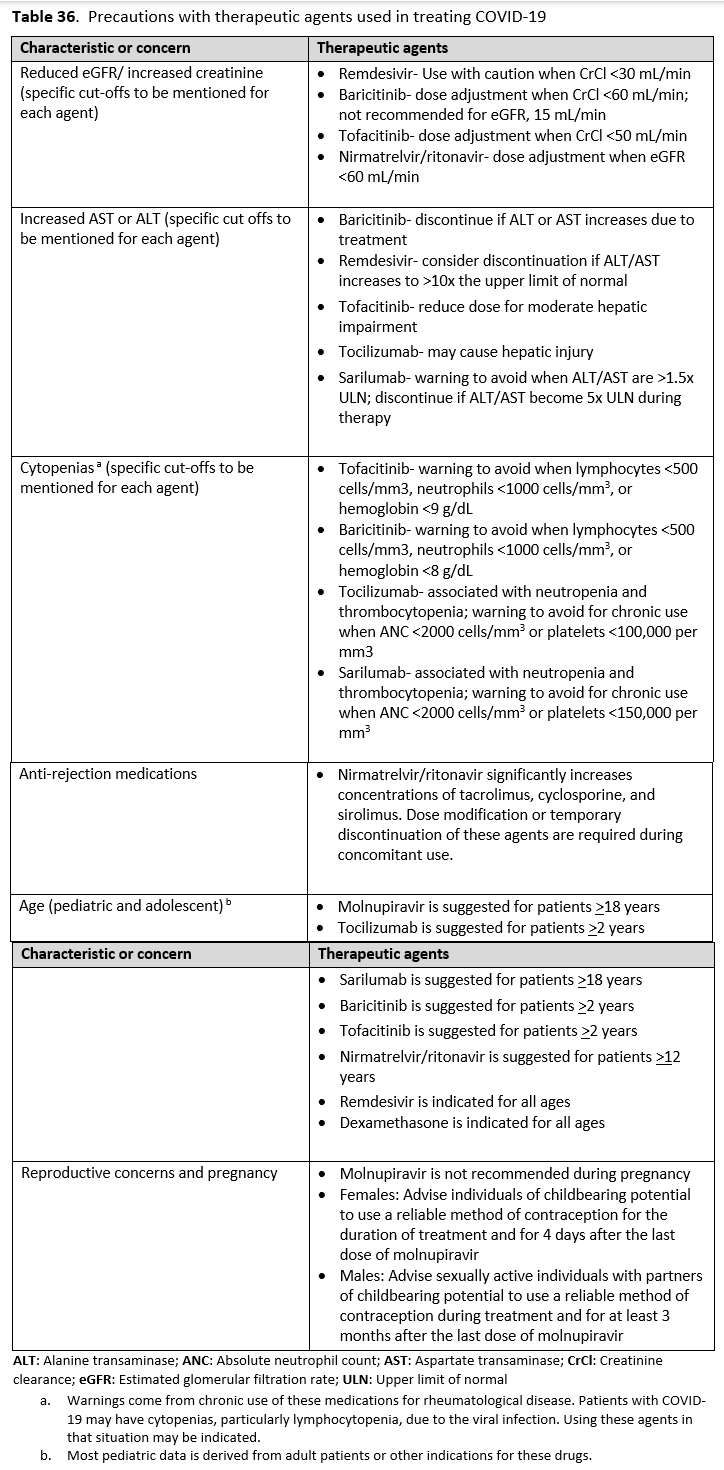

Pharmacologic treatment of mild-to-moderate COVID-19 with risk factors for progression
COVID-19 is considered mild when there are clinical features suggestive of upper res-piratory tract involvement without features of lung or other end organ involvement. Moderate COVID-19 is pulmonary involvement with no hypoxia. Most patients improve with supportive care at this stage, but patients with risk factors can progress to more severe or critical disease or death; such individuals may benefit from pharmacotherapies. There are no validated clinical prediction rules or risk calculators, but the FDA EUA and CDC mention a few of these risk fac-tors to consider for treatment with anti-SARS-CoV-2 antibodies [276]. More research is needed to identify prediction instruments and determinants that both increase or decrease the risk of severe disease and how potentially protective factors influence risk stratification. Most of these treatments are effective only when given early, within 5-7 days of symptom onset.
Patients who have these risk factors should be offered treatment with nirma-trelvir/ritonavir for 5 days (oral) or remdesivir for 3 days (intravenous). If these agents are not available or cannot be used then consider molnupiravir for 5 days (oral) or, if immunocompro-mised, high-titer convalescent plasma (intravenous) with activity against circulating variant. Convalescent plasma obtained from people who have recovered from COVID-19 due to Omi-cron and have been vaccinated is expected to be active against Omicron.
arenteral anti-SARS-CoV-2 monoclonal antibodies can be used to treat if the circulating SARS CoV-2 variants in that region are susceptible to the specific agent, given trials have shown a reduction in the need for hospitalizations, ER visits or medically attended visit. At present (2/2/2023) a significant proportion of the circulating SARS CoV-2 variants in the US are not sus-ceptible to most of the neutralizing antibodies. There are no neutralizing antibodies that are currently (2/2/2023) authorized or approved by US FDA.
There are logistical issues related to administration of parenteral agents in ambulatory settings which may preclude their use. Oral antivirals like nirmatrelvir/ritonavir and mol-nupiravir have an advantage as they are easy to prescribe in outpatient settings, but there are significant limitations and unique considerations that need to be addressed by providers, which might be a barrier to their timely use. In the United States, many of the antiviral treatments do not have authorization for use in patients admitted to the hospital for mild-to-moderate COVID-19 but can be used if they are admitted for another reason and found to have mild-to-moderate COVID-19. We do not recommend using hydroxychloroquine, azithromycin, or lop-inavir/ritonavir as trials have shown no evidence of benefit.
We recommend against the use of ivermectin outside of the context of a clinical trial given the low certainty of evidence for its benefit. We also do not recommend the use of sys-temic corticosteroids in mild-to-moderate COVID-19. Though the RECOVERY trial was complet-ed in hospitalized patients and not ambulatory patients, it demonstrated a trend to increase mortality when used in patients with mild-to-moderate COVID-19 (relative risk 1.22; 95% CI 0.86, 1.75) [95].
Pharmacologic treatment of severe COVID-19
Patients with severe COVID-19 are those whose infection has pulmonary involvement resulting in hypoxia while breathing room air and/or needing treatment with low flow oxygen. Most existing criteria for trials consider either a SpO2 level less than 94% or 90% or tachypnea (respiratory rate >30 breaths per minute) as severe COVID-19. Clinical judgment of individual cases should supplement these criteria.
Corticosteroids, especially dexamethasone, has demonstrated a mortality benefit are recommended as the cornerstone of therapy in severe COVID-19. Remdesivir may be consid-ered as it has shown to decrease time to recovery or discharge, though it has not been shown to improve mortality [32, 159].
The IL-6 inhibitors tocilizumab and sarilumab [111, 277] and JAK inhibitors baricitinib and tofacitinib [182] have shown a benefit in severe, but non-critical COVID-19 when used with corticosteroids. The trials did not identify specific sub-populations of patients with severe COVID-19 already being treated with corticosteroids who would benefit most with additional treatment with IL-6 or JAK inhibitors. We recommend using either IL-6 inhibitors or JAK inhibi-tors (baricitinib preferred over tofacitinib) in those patients who have elevated inflammatory markers like CRP and progressive severe COVID-19. Since there is greater supportive data for tocilizumab and baricitinib we recommend them preferentially over sarilumab and tofacitinib, though the latter agents are suitable alternatives if the former are not available. We do not recommend using hydroxychloroquine, azithromycin, lopinavir/ritonavir, or convalescent plas-ma as trials have not shown a benefit in patients with severe disease. We also recommend against the use of ivermectin outside of the context of a clinical trial given the low certainty of evidence for its benefit.
Pharmacologic treatment of critically ill COVID-19 requiring non-invasive ventilation or oxygen by high-flow nasal cannula
Critically ill patients with COVID-19 need more ventilatory or oxygenation support either with high-flow oxygen or with noninvasive ventilation. High-flow oxygen therapy involves deliv-ery of oxygen via special devices at rates greater than those possible via a simple nasal canula.
We strongly recommend systemic corticosteroids in critically ill patients with COVID-19 as they have shown a mortality benefit in this population (OR: 0.66; 95% CI: 0.54; 0.82) [79]. In critically ill patients, dexamethasone 6mg/day is preferred but doses up to 20 mg/day can be used if indicated for other reasons. Hydrocortisone 50 mg IV Q6 hours is an alternative that has also been studied. Methylprednisolone and prednisone have less supporting data but are rea-sonable pharmacologic alternatives at equipotent doses. In addition to corticosteroids, we rec-ommend using either IL-6 inhibitors (tocilizumab preferred over sarilumab) or JAK inhibitors (baricitinib preferred over tofacitinib) in patients who have elevated inflammatory markers (e.g., CRP), which most critically ill COVID-19 patients have. The trials done so far have not identified specific sub-populations of critically ill patients already being treated with cortico-steroids who would benefit with additional treatment with IL-6 or JAK inhibitors. We do not rec-ommend remdesivir since it has not shown a benefit in this sub-population [159].
Pharmacologic treatment of critically ill COVID-19, needing invasive mechanical ventilation or ECMO
Patients who are critically ill with COVID-19 pulmonary disease and dysfunction needing significant ventilatory support with invasive mechanical ventilation or ECMO have the highest risk of mortality. Pharmacologically, we recommend treating them similarly to those on non-invasive ventilation or high-flow nasal cannula. Corticosteroids are strongly recommended in this category of critically ill patients as trials have demonstrated a mortality benefit [79]. In ad-dition to steroids, the panel recommends using either IL-6 inhibitors (tocilizumab is preferred over sarilumab) in critically ill patients who have elevated inflammatory markers like CRP. In situations where IL-6 inhibitors are not available, baricitinib can be used in mechanically venti-lated patients as a small trial showed a mortality benefit in this population [278]. Most other COVID-19 therapies studied in other severities have either not demonstrated benefit or not been studied in this population.
Bacterial Co-Infections and Antibiotic Use
Patients with COVID-19 often present with viral pneumonia with accompanying febrile illness and respiratory symptoms. Differential diagnoses may include bacterial pneumonia, for which antibiotics are prescribed. Concerns also exist for bacterial superinfections in hospital-ized patients during the course of illness. Studies reported to date mainly describe antibiotic use during the early phase of the COVID-19 pandemic and consistently report high percentages of antibiotic use worldwide (58-95%) [1, 279-285]. One registry of 150 Spanish hospitals found that over 75% of patients received antibiotics, but diagnosis in the early months of the pandemic was a predictor of inappropriate antibiotic use. Antibiotic use was associated with adverse drug reactions [286].
Data reporting co-infection in patients presenting with COVID-19 for care has mostly fo-cused on patients receiving care in hospitals. As more studies have become available, they can be grouped into those describing co-infection at the diagnosis of COVID-19, those describing the treatment of superinfections during the course of COVID-19 infection, those that report both, and those that do not distinguish between these types of infections. The latter are not discussed here.
Despite the majority of patients with COVID-19 being treated with antibiotics on admis-sion early in the pandemic, existing studies have found bacterial co-infections to be uncommon. Vaughn and colleagues evaluated a random cohort of patients with COVID-19 across 38 hospi-tals in Michigan. Of the 1705 patients included, only 3.5% had a bacterial co-infection, though 59.5% received antibacterial drugs [283]. A cohort of 1016 patients with COVID-19 across five Maryland hospitals found bacterial co-infection in only 1.2% [287]. A meta-analysis including 3338 patients in 24 studies reported bacterial co-infection in 3.5% [288]. Smaller studies had congruent reports, ranging from 3.1 to 4% [289-291]. A study of 64,961 COVID-19 patients in the Premier Healthcare Database is an outlier, reporting bacterial co-infections in 18.5% of in-fections between April and June 2020, but this relied on ICD-10 codes and not microbiological diagnoses. Urinary tract infections were most reported [292].
Studies describing superinfections that developed in patients with COVID-19 are more heterogeneous. Studies that describe the incidence of superinfection in entire hospitalized co-horts of COVID-19 report incidences of superinfection of 4.2 to 21% [288, 291, 293]. Small stud-ies of patients requiring mechanical ventilation and with COVID-19-associated ARDS reported superinfections in 44.4% and 27.7% of patients, respectively [294, 295].
The apparent discordance between bacterial and fungal co-infection in patients with COVID-19 at presentation and the use of antibacterial therapy has potential negative effects, namely in antimicrobial resistance. Several studies have attempted to differentiate patients with and without concomitant bacterial infections using laboratory data. The use of procalciton-in in a group of hospitals was not effective as tool to encourage antibiotic discontinuation com-pared to clinical judgment [296]. Mason and colleagues compared hospitalized cohorts of 619 patients with COVID-19 and 106 with community-acquired bacterial pneumonia (CABP) to de-termine if inflammatory markers could be used to rule out bacterial co-infection [297]. They found marked differences in white blood cell counts between groups (6.78 COVID-19 vs. 12.48 CABP), and that CRP declined in 48-72 hours with antibiotic therapy in the CABP cohort but not the COVID-19 group, suggesting that these can be used to guide antibiotic discontinuation when initiated empirically in COVID-19 patients. Initiating and continuing empiric antibiotics at the time of admission may lead to superinfections that are antibiotic resistant; one study found an-tibiotic use in the first two days of admission for COVID-19 to be a risk factor for superinfection [293]. Immunomodulatory therapies are recommended for many patients with severe and criti-cal illness from COVID-19, including corticosteroids, IL-6 antagonists, JAK inhibitors, and others [298]. Most of the prospective studies that support these recommendations have not reported higher rates of infection in patients receiving immunomodulators, but follow-up is limited in most cases and late infections may be missed.
Pediatric Considerations for Treatment of SARS-CoV-2 Infection and Multisystem Inflammatory Syndrome in Children
Acute SARS-CoV-2 Infection in Children
Clinical presentation
Case [299, 300] and hospitalization rates [301] from SARS-CoV-2 infection in children are lower than in adults, and asymptomatic infection is more common [302, 303]. However, infection can lead to significant illness and even death in children [304-306]. Clinical presenta-tions of infection can be non-specific, and may more frequently include fever alone and/or gas-trointestinal symptoms [307] than in adults. Children are also capable of transmitting disease to others [308].
Factors which lead to severe illness in children with SARS-CoV-2 infection are less well-defined than in adults. Comorbidities including medically complex conditions (including certain genetic disorders, neurologic diseases, and cancer) [309], type 1 diabetes, complex congenital heart disease, and obesity have all been associated with a higher risk of hospitalization and ICU admission in children [306, 310-312].
Management
Remdesivir
The studies involving the use of remdesivir in hospitalized patients with COVID-19 (recommendations 15-17) [32, 159-161, 313] have generally focused on individuals over age 18 years. Two trials included children over 12 years [161, 313], but did not separately report the number or outcomes (including adverse events) of participants under 18 years. Nevertheless, remdesivir is commonly used and recommended by expert panels [314] of pediatric ID specialists in hospitalized children with SARS-CoV-2 infection, and reports suggest low adverse event rates [162, 315]. An ongoing phase II/III open label study in children (the “CARAVAN” trial) [163] has not yet reported their results in the peer-reviewed literature [316]. Recent studies of outpatient remdesivir treatment in individuals at high risk for progression support its use in pediatric patients down to 3.5 kg of body weight.
Corticosteroids
Dexamethasone and other corticosteroids are recommended in certain hospitalized patients with COVID-19 (recommendations 7-9). The studies informing these recommendations [79, 95] either did not include children or did not separately report the number or outcomes (including adverse events) of participants under 18 [95] years. Corticosteroid use is nevertheless common in hospitalized children with COVID-19 [311], and there is reason to believe that the risk benefit ratio would be similar in children and adults.
IL-6 blockade
Tocilizumab or sarilumab is suggested for use in treatment of COVID-19 in certain situations (recommendations 11-12). Of the studies informing the recommendations for tocilizumab [110, 111, 113-116, 317, 318], only two [110, 111] did not specifically exclude children under 18 years from enrolling. The RECOVERY trial included children, but results from those in the tocilizumab arm of the trial have not yet been reported. Hermine et al. did not specifically exclude children, but results in children were not separately reported either.
Three of the four studies used to inform the recommendations for sarilumab excluded children from participation [117, 118, 317]. The pre-print network meta-analysis of 18 RCTs of IL-6 inhibitors included some studies that enrolled children, but results in children were not separately reported.
There are several publications reporting on cohorts of children with COVID-19 who received treatment with tocilizumab [315, 319-321]. Although there have been no clear contraindications to using IL-6 inhibitors in children based on these reports more studies in children are needed to determine whether the criteria for their pediatric use would be similar to those in adults.
JAK inhibitors
Baricitinib is suggested for use in treating certain hospitalized patients with COVID-19 (recommendations 21-22). However, the studies which inform these recommendations did not include children [176, 181, 182, 278]. Although the EUA for use of baricitinib in treatment of COVID-19 extends to children over 2 years of age [322], baricitinib does not have an FDA indication for treatment of other conditions in children, and there are only limited published pediatric pharmacokinetic data [323]. A pediatric safety and pharmacokinetic study on baricitinib use in children with COVID-19 is now recruiting [324].
Tofacitinib is also suggested for use in treating certain hospitalized patients with COVID-19 (recommendation 23). As with baricitinib, the trial informing this recommendation did not include children [185]. Tofacitinib is used in children over age 2 and over 10 kg for treatment of polyarticular juvenile idiopathic arthritis when they have had an inadequate response or intolerance to one or more tumor necrosis factor inhibitors [325]. There are no currently open trials studying tofacitinib for treatment of COVID-19 in children.
Oral antivirals
Two new antiviral agents have been issued an EUA and include: nirmatrelvir/ritonavir and molnupiravir. Nirmatrelvir/ritonavir is not authorized in children younger than 12 years of age and weighing less than 40 kg [326]. However, there have been no safety or effectiveness studies in pediatric patients. Molnupiravir is not recommended for use in children due to animal studies that suggest effects on bone and cartilage growth.
Monoclonal antibodies
At earlier stages in the pandemic, neutralizing monoclonal antibodies directed against the spike protein of SARS-CoV-2 have been used for pre- and post-exposure prophylaxis and treatment of individuals exposed to or infected with SARS-CoV-2 who are at high risk of progression to severe disease, but emergence of variants with in vitro reductions in susceptibility to these antibodies has left no available products in the United States. As noted previously, use of these products may be considered in areas of the world where a significant proportion of circulating variants retain susceptibility, taking into account the predicted relative benefits of the anti-SARS CoV-2 neutralizing antibody product compared with alternative antiviral therapies. In children, clinicians should also consider limitations in the age ranges and minimum body weight in which these products have been studied and should note that risk factors for progression to severe illness in children are less well-defined than in adults. Although risk-benefit ratios for the use of SARS-CoV-2 monoclonal antibodies are likely similar between children and adults, pediatric-specific data are limited or lacking for all neutralizing monoclonal antibody products.
Treatments not recommended for use
As noted in other sections of this document, several interventions have been tested in adult populations and not found to have clinical benefit. This has led to recommendations against the routine use of hydroxychloroquine, lopinavir/ritonavir, inpatient convalescent plasma, and famotidine. Although the studies informing these recommendations largely excluded children with acute infection, the experience in adult patients suggests that these drugs would not be expected to have benefit in treatment of children with similar disease characteristics.
Multisystem Inflammatory Syndrome in Children
Clinical presentation
Multisystem inflammatory syndrome in children (MIS-C), also called Pediatric Inflammatory Multisystem Syndrome temporally associated with COVID-19 (PIMS-TS), is a rare acute inflammatory syndrome reported in children several weeks following acute SARS-CoV-2 infection. Case definitions for this syndrome were derived after reports of critically ill children presenting with fever, rash, conjunctivitis, abdominal complaints, shock, and significant cardiac dysfunction in the setting of recent SARS-CoV-2 infection [327-339] (Table 38). Incidence of MIS-C is higher in Black, Hispanic or Latinx, and Asian or Pacific Islander children than in Caucasian children and most common among children between 6 and 10 years of age [340, 341]. Epidemiologic data showing clusters of MIS-C cases following peaks of positive SARS-CoV-2 test rates by 2-5 weeks [342] support that the syndrome results from a delayed immunologic response to the infection.
Management
Once the diagnosis of MIS-C has been made, immunomodulatory medications are the mainstay of therapy. Although trials are lacking to demonstrate the superiority of any given ap-proach, intravenous immunoglobulin (IVIG) and systemic steroids are frequent initial choices [336, 343]. Studies comparing outcomes after initial treatment using IVIG alone, steroids alone, or a combination of IVIG and steroids have come to differing conclusions on their relative im-portance in treatment. The combination of both has been reported to lead to faster and more sustained resolution of fever than IVIG alone [344]. Biologic treatments including anakinra, in-fliximab, or tocilizumab have also been used in refractory cases [343, 345-347], though data are limited to inform the choice among these interventions or those patients who would benefit most. Despite these limitations, overall outcomes of children with MIS-C have been generally good with few fatalities reported [339, 348].
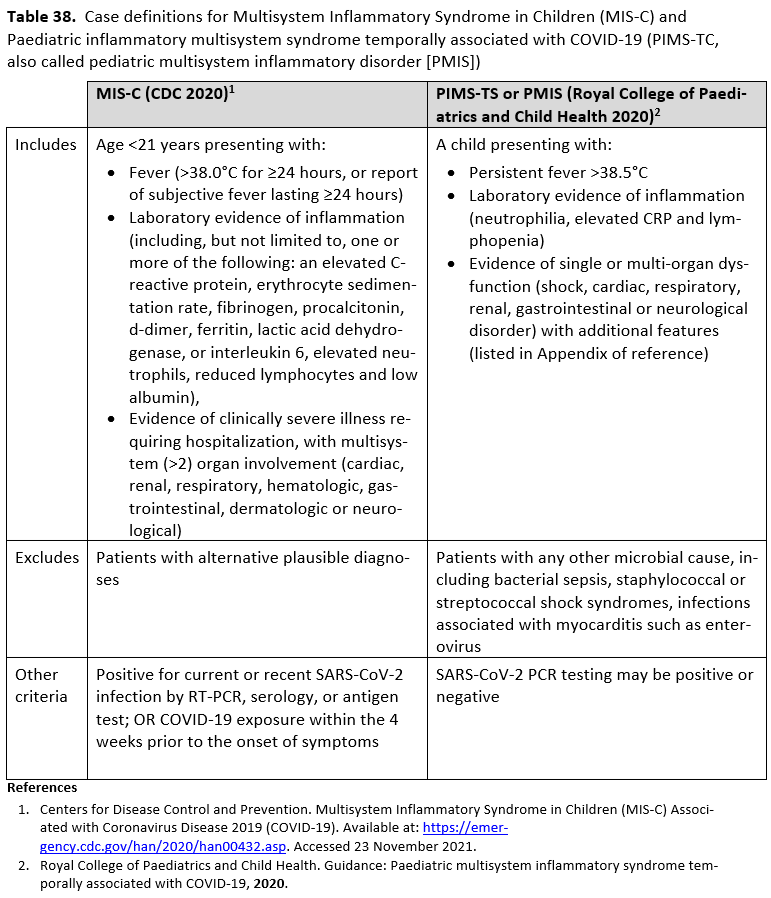
Notes
Acknowledgement
The expert panel thanks the Infectious Diseases Society of America for supporting guideline development, and specifically Imani Amponsah, Genet Demisashi, Jon Heald, Hannah Rehm, Sheila Tynes, and Dana Wollins for their continual support and guidance the last two years in developing and maintaining the living rapid guidelines. This guideline would have been impossible without their help.
Financial Support
This project was funded in part by a cooperative agreement with the Centers for Disease Control and Prevention (CDC) (grant number 6 NU50CK000477-04-01). The CDC is an agency within the Department of Health and Human Services (HHS). The contents of this guideline do not necessarily represent the policy of CDC or HHS and should not be considered an endorsement by the Federal Government.
COI Summary
The following list is a reflection of what has been reported to IDSA. To provide thorough transparency, IDSA requires full disclosure of all relationships, regardless of relevancy to the guideline topic. Evaluation of such relationships as potential conflicts of interest is determined by a review process which includes assessment by the Board of Directors liaison to the Standards and Practice Guidelines Committee and, if necessary, the Conflicts of Interest and Ethics Committee. The assessment of disclosed relationships for possible COI is based on the relative weight of the financial relationship (i.e., monetary amount) and the relevance of the relationship (i.e., the degree to which an association might reasonably be interpreted by an independent observer as related to the topic or recommendation of consideration). The reader of these guidelines should be mindful of this when the list of disclosures is reviewed. L.B. receives research funding from the National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID), Bill and Melinda Gates Foundation, Wellcome Trust, and Harvard Medical School; serves as chair of the Antimicrobial Drug Advisory Committee of the Food and Drug Administration; and is involved in HIV and COVID-19 vaccine clinical trials conducted in collaboration with the NIH, HIV Vaccine Trials Network, COVID Vaccine Prevention Network, International AIDS Vaccine Initiative, Crucell/Janssen Pharmaceuticals, Moderna, Military HIV Research Program, Bill and Melinda Gates Foundation, and the Ragon Institute. A.B. received honorarium from the Institute for Clinical and Economic Review. V.C. receives research funding from the Health and Medical Research Fund; serves on the Research Committee of the Society for Healthcare Epidemiology of America (SHEA); and serves on the international editorial boards for the Journal of Hospital Infection, Infection Prevention in Practice, and Antimicrobial Stewardship and Healthcare Epidemiology. K.E. serves as a scientific advisor for Merck, Bionet, IBM, Sanofi, X4 Pharmaceuticals, Inc., Seqirus, Inc., Moderna, Inc., GSK plc, Roche, and Pfizer; and receives research funding from the Centers for Disease Control and Prevention and the NIH. J.G. serves in an advisory role for Qpex, Shionogi, and Merck; receives research funding from Merck; previously served in an advisory role for Accelerate Diagnostics, Achaogen, Astellas Pharma, Melinta Therapeutics, Nabriva Therapeutics, Paratek Pharma, scPharmaceuticals, Spero Therapeutics, and Tetraphase Pharmaceuticals; and previously served on the speakers bureau for Astellas Pharma, Melinta Therapeutics, Merck, and Shionogi. R.G. serves as a panel member on the NIH COVID-19 Treatment Guidelines Panel; serves as the immediate Past Chair for the HIV Medicine Association; receives research funding from the NIH; and has served on the scientific advisory board for Gilead Sciences, Inc., and Merck. W.J.M. serves in an advisory role for Seqirus, Inc.; receives research funding from Ansun Biopharma, Astellas Pharma, AstraZeneca, Eli Lilly and Company, Enanta Pharmaceuticals, Gilead Sciences, Janssen Pharmaceuticals, Karius, Melinta Therapeutics, Merck, Moderna, Nabriva Therapeutics, Paratek Pharma, Pfizer, Roche, and Tetraphase Pharmaceuticals; and has previously received research funding from Abbott Laboratories. M.H.M receives research funding from the Agency for Healthcare Research and Quality, the Endocrine Society, and the Society for Vascular Surgery; serves as a Board member for the Evidence Foundation; has received research funding from the American Society of Hematology and the World Health Organization (WHO); and has served as a guideline methodologist for the WHO. R.A.M. receives research funding from the NIH, the WHO, the American College of Rheumatology, the American Society of Hematology, and Bohringer Ingelheim; serves as Chair of the Midwest Comparative Effectiveness Public Advisory Council of the Institute for Clinical and Economic Review (ICER); serves on the Methods Committee for Kidney Disease Improving Global Outcomes Work Group; serves on the Clinical Guidelines Committee for the Canadian Society of Nephrology; and previously served on the Clinical Guidelines Committee for the American College of Physicians (ACP). M.M.N. co-chairs the Pediatric Infectious Diseases Society COVID-19 Therapies Task Force, will receive support to attend as a speaker the American Academy of Pediatrics National Conference & Exhibition in October 2022, and has received research funding from Gilead Sciences. J.C.O. serves as an advisor for Bates College; holds stocks in Doximity, Inc.; receives research funding from the MITRE Corporation and Nference, Inc.; and serves on committees for the Society for Critical Care Medicine, SHEA, and University Lake School. R.W.S. served in an advisory role for GSK plc and Gilead Sciences. S.S. serves in advisory roles for Amplyx Pharmaceuticals, Inc., ReViral Ltd., Adamis Pharmaceuticals, and Immunome; holds stocks in Immunome; receives research funding from Ansun BioPharma, Zeteo Tech, Inc., F2G, Emergent Biosolutions, Shionogi, Shire (now Takeda), Cidara Therapeutics, U.S. Department of Defense (Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense), Defense Health Agency, Bloomberg Philanthropies, the State of Maryland, NIH/NIAID, NIH National Center for Advancing Translational Sciences, Mental Wellness Foundation, Moriah Fund, Octopharma, HealthNetwork Foundation, Shear Family Foundation, Johns Hopkins University, and Mayo Clinic; serves as the Governor of the ACP; has received research funding from the University of Nebraska; and has served as an advisor for Janssen Pharmaceuticals, Acidophil, LLC, Adagio Therapeutics, Inc., Celltrion Healthcare, and Intermountain Health. A.H.S. receives research funding from the U.S. Department of Veterans Affairs. S.S. serves on guideline panels for the American Gastroenterological Association (AGA) and receives research funding from the Department of Veterans Affairs Evidence Synthesis Program. Y.F.Y. receives honoraria from the Evidence Foundation for evidence reviews and teaching, the AGA for evidence reviews, and ICER for committee meetings; serves as a Director for the Evidence Foundation and for the U.S. GRADE Network; and served on an Independent Appraisal Committee for ICER. All other authors: no disclosures reported. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
IDSA Disclaimer
It is important to realize that guidelines cannot always account for individual variation among patients. They are assessments of current scientific and clinical information provided as an educational service; are not continually updated and may not reflect the most recent evidence (new evidence may emerge between the time information is developed and when it is published or read); should not be considered inclusive of all proper treatments methods of care, or as a statement of the standard of care; do not mandate any particular course of medical care; and are not intended to supplant physician judgment with respect to particular patients or special clinical situations. Whether and the extent to which to follow guidelines is voluntary, with the ultimate determination regarding their application to be made by the physician in the light of each patient’s individual circumstances. While IDSA makes every effort to present accurate, complete, and reliable information, these guidelines are presented “as is” without any warranty, either express or implied. IDSA (and its officers, directors, members, employees, and agents) assume no responsibility for any loss, damage, or claim with respect to any liabilities, including direct, special, indirect, or consequential damages, incurred in connection with these guidelines or reliance on the information presented.
The guidelines represent the proprietary and copyrighted property of IDSA. Copyright 2023 Infectious Diseases Society of America. All rights reserved. No part of these guidelines may be reproduced, distributed, or transmitted in any form or by any means, including photocopying, recording, or other electronic or mechanical methods, without the prior written permission of IDSA. Permission is granted to physicians and health care providers solely to copy and use the guidelines in their professional practices and clinical decision-making. No license or permission is granted to any person or entity, and prior written authorization by IDSA is required, to sell, distribute, or modify the guidelines, or to make derivative works of or incorporate the guidelines into any product, including but not limited to clinical decision support software or any other software product. Except for the permission granted above, any person or entity desiring to use the guidelines in any way must contact IDSA for approval in accordance with the terms and conditions of third-party use, in particular any use of the guidelines in any software product.
References
1. Guan WJ, Ni ZY, Hu Y, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med 2020; 382(18): 1708-20.
2. World Health Organization. Coronavirus disease 2019 (COVID-19) Situation Report - 75. Geneva: World Health Organization, 2020 4 April.
3. Wu Z, McGoogan JM. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020.
4. Morgan RL, Florez I, Falavigna M, et al. Development of rapid guidelines: 3. GIN-McMaster Guideline Development Checklist extension for rapid recommendations. Health Res Policy Syst 2018; 16(1): 63.
5. Guyatt GH, Oxman AD, Kunz R, et al. GRADE guidelines: 2. Framing the question and deciding on important outcomes. J Clin Epidemiol 2011; 64(4): 395-400.
6. U.S. Food and Drug Administration. What is a Serious Adverse Event? Available at: https://www.fda.gov/safety/reporting-serious-problems-fda/what-serious-adverse-event. Accessed 19 June 2020.
7. National Institute for Health and Care Excellence. Scoping. Interim process and methods for developing rapid guidelines on COVID-19 (PMG35). London: National Institute for Health and Care Excellence, 2020.
8. Wallace BC, Dahabreh IJ, Trikalinos TA, Lau J, Trow P, Schmid CH. Closing the gap between methodologists and end-users: R as a computational back-end. J Stat Softw 2012; 49(5): 1-15.
9. Higgins JPT, Thomas J, Chandler J, et al. Cochrane Handbook for Systematic Reviews of Interventions. 2 ed. Chichester (UK): John Wiley & Sons, 2019.
10. Sterne JA, Hernan MA, Reeves BC, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016; 355: i4919.
11. Guyatt G, Oxman AD, Akl EA, et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol 2011; 64(4): 383-94.
12. GRADEpro GDT. GRADEpro Guideline Development Tool [Software]. McMaster University, 2015 (developed by Evidence Prime, Inc.). Available at: https://gradepro.org/.
13. Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol 2012; 42(2): 145-53.
14. Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun 2004; 323(1): 264-8.
15. Dyall J, Coleman CM, Hart BJ, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother 2014; 58(8): 4885-93.
16. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 2020; 30(3): 269-71.
17. Yao X, Ye F, Zhang M, et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin Infect Dis 2020.
18. Vincent MJ, Bergeron E, Benjannet S, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J 2005; 2: 69.
19. Gautret P, Lagier JC, Parola P, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J Antimicrob Agents 2020; 56(1): 105949.
20. Li C, Zu S, Deng YQ, et al. Azithromycin Protects against Zika virus Infection by Upregulating virus-induced Type I and III Interferon Responses. Antimicrob Agents Chemother 2019.
21. Kouznetsova J, Sun W, Martinez-Romero C, et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect 2014; 3(12): e84.
22. Gielen V, Johnston SL, Edwards MR. Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur Respir J 2010; 36(3): 646-54.
23. Tyteca D, Van Der Smissen P, Mettlen M, et al. Azithromycin, a lysosomotropic antibiotic, has distinct effects on fluid-phase and receptor-mediated endocytosis, but does not impair phagocytosis in J774 macrophages. Exp Cell Res 2002; 281(1): 86-100.
24. Menzel M, Akbarshahi H, Bjermer L, Uller L. Azithromycin induces anti-viral effects in cultured bronchial epithelial cells from COPD patients. Sci Rep 2016; 6: 28698.
25. Takizawa H, Desaki M, Ohtoshi T, et al. Erythromycin suppresses interleukin 6 expression by human bronchial epithelial cells: a potential mechanism of its anti-inflammatory action. Biochem Biophys Res Commun 1995; 210(3): 781-6.
26. Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54(1): 21-8.
27. Arshad S, Kilgore P, Chaudhry ZS, et al. Treatment with hydroxychloroquine, azithromycin, and combination in patients hospitalized with COVID-19. Int J Infect Dis 2020; 97: 396-403.
28. Cavalcanti AB, Zampieri FG, Rosa RG, et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N Engl J Med 2020; 383: 2041-52.
29. Horby P, Mafham M, Linsell L, et al. Effect of Hydroxychloroquine in Hospitalized Patients with COVID-19: Preliminary results from a multi-centre, randomized, controlled trial. medRxiv 2020: Available at: https://doi.org/10.1101/2020.07.15.20151852 [Preprint 15 July 2020].
30. Chen J, Liu D, Liu L, et al. A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19. Journal of Zhejiang University (Medical Sciences) 2020; 49(2): 215-9.
31. Chen Z, Hu J, Zhang Z, et al. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. medRxiv 2020: Available at: https://doi.org/10.1101/2020.03.22.20040758 [Preprint 10 April 2020].
32. WHO Solidarity Trial Consortium, Pan H, Peto R, et al. Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N Engl J Med 2021; 384(6): 497-511.
33. Self WH, Semler MW, Leither LM, et al. Effect of Hydroxychloroquine on Clinical Status at 14 Days in Hospitalized Patients With COVID-19: A Randomized Clinical Trial. JAMA 2020; 324(21): 2165-76.
34. Ulrich RJ, Troxel AB, Carmody E, et al. Treating COVID-19 With Hydroxychloroquine (TEACH): A Multicenter, Double-Blind Randomized Controlled Trial in Hospitalized Patients. Open Forum Infect Dis 2020; 7(10): ofaa446.
35. Tang W, Cao Z, Han M, et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ 2020; 369: m1849.
36. Geleris J, Sun Y, Platt J, et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N Engl J Med 2020; 382(25): 2411-8.
37. Magagnoli J, Narendran S, Pereira F, et al. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with Covid-19. medRxiv 2020: Available at: https://doi.org/10.1101/2020.04.16.20065920 [Preprint 23 April 2020].
38. Mahevas M, Tran V-T, Roumier M, et al. No evidence of clinical efficacy of hydroxychloroquine in patients hospitalized for COVID-19 infection with oxygen requirement: results of a study using routinely collected data to emulate a target trial. medRxiv 2020: Available at: https://doi.org/10.1101/2020.04.10.20060699 [Preprint 14 April 2020].
39. Rosenberg ES, Dufort EM, Udo T, et al. Association of treatment with hydroxychloroquine or azithromycin with in-hospital mortality in patients with COVID-19 in New York state. JAMA 2020; 323(24): 2493-502.
40. Yu B, Li C, Chen P, et al. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci China Life Sci 2020; 63(10): 1515-21.
41. Ip A, Berry DA, Hansen E, et al. Hydroxychloroquine and Tocilizumab Therapy in COVID-19 Patients-An Observational Study. medRxiv 2020: Available at: https://doi.org/10.1101/2020.05.21.20109207 [Preprint 25 May 2020].
42. Cipriani A, Zorzi A, Ceccato D, et al. Arrhythmic profile and 24-hour QT interval variability in COVID-19 patients treated with hydroxychloroquine and azithromycin. Int J Cardiol 2020; 316: 280-4.
43. Molina JM, Delaugerre C, Goff J, et al. No Evidence of Rapid Antiviral Clearance or Clinical Benefit with the Combination of Hydroxychloroquine and Azithromycin in Patients with Severe COVID-19 Infection. Médecine et Maladies Infectieuses 2020; 50(4): 384.
44. Gautret P, Lagier JC, Parola P, et al. Clinical and microbiological effect of a combination of hydroxychloroquine and azithromycin in 80 COVID-19 patients with at least a six-day follow up: A pilot observational study. Travel Med Infect Dis 2020; 34: 101663.
45. Chorin E, Dai M, Shulman E, et al. The QT Interval in Patients with SARS-CoV-2 Infection Treated with Hydroxychloroquine/Azithromycin. medRxiv 2020: Available at: https://doi.org/10.1101/2020.04.02.20047050 [Preprint 3 April 2020].
46. Wang C, Fortin PR, Li Y, Panaritis T, Gans M, Esdaile JM. Discontinuation of antimalarial drugs in systemic lupus erythematosus. J Rheumatol 1999; 26(4): 808-15.
47. Youngster I, Arcavi L, Schechmaster R, et al. Medications and glucose-6-phosphate dehydrogenase deficiency: an evidence-based review. Drug Saf 2010; 33(9): 713-26.
48. Mohammad S, Clowse MEB, Eudy AM, Criscione-Schreiber LG. Examination of Hydroxychloroquine Use and Hemolytic Anemia in G6PDH-Deficient Patients. Arthritis Care Res (Hoboken) 2018; 70(3): 481-5.
49. Beauverd Y, Adam Y, Assouline B, Samii K. COVID-19 infection and treatment with hydroxychloroquine cause severe haemolysis crisis in a patient with glucose-6-phosphate dehydrogenase deficiency. Eur J Haematol 2020.
50. Kuipers MT, van Zwieten R, Heijmans J, et al. G6PD deficiency-associated hemolysis and methemoglobinemia in a COVID-19 patient treated with chloroquine. Am J Hematol 2020.
51. Maillart E, Leemans S, Van Noten H, et al. A case report of serious haemolysis in a glucose-6-phosphate dehydrogenase-deficient COVID-19 patient receiving hydroxychloroquine. Infect Dis (Lond) 2020: 1-3.
52. Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF. Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 2015; 23(5): 231-69.
53. Morgan ND, Patel SV, Dvorkina O. Suspected hydroxychloroquine-associated QT-interval prolongation in a patient with systemic lupus erythematosus. J Clin Rheumatol 2013; 19(5): 286-8.
54. Chen CY, Wang FL, Lin CC. Chronic hydroxychloroquine use associated with QT prolongation and refractory ventricular arrhythmia. Clin Toxicol (Phila) 2006; 44(2): 173-5.
55. Yelve K, Phatak S, Patil MA, Pazare AR. Syncope in a patient being treated for hepatic and intestinal amoebiasis. BMJ Case Rep 2012; 2012: bcr2012006687.
56. Stas P, Faes D, Noyens P. Conduction disorder and QT prolongation secondary to long-term treatment with chloroquine. Int J Cardiol 2008; 127(2): e80-2.
57. Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366(20): 1881-90.
58. von Rosensteil NA, Adam D. Macrolide antibacterials. Drug interactions of clinical significance. Drug Saf 1995; 13(2): 105-22.
59. Barnabas RV, Brown ER, Bershteyn A, et al. Hydroxychloroquine as Postexposure Prophylaxis to Prevent Severe Acute Respiratory Syndrome Coronavirus 2 Infection : A Randomized Trial. Ann Intern Med 2021; 174(3): 344-52.
60. Boulware DR, Pullen MF, Bangdiwala AS, et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med 2020; 383(6): 517-25.
61. Mitja O, Corbacho-Monne M, Ubals M, et al. A Cluster-Randomized Trial of Hydroxychloroquine for Prevention of Covid-19. N Engl J Med 2021; 384(5): 417-27.
62. Chen F, Chan KH, Jiang Y, et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J Clin Virol 2004; 31(1): 69-75.
63. Wu CY, Jan JT, Ma SH, et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci U S A 2004; 101(27): 10012-7.
64. Chan JF, Yao Y, Yeung ML, et al. Treatment With Lopinavir/Ritonavir or Interferon-beta1b Improves Outcome of MERS-CoV Infection in a Nonhuman Primate Model of Common Marmoset. J Infect Dis 2015; 212(12): 1904-13.
65. Chu CM, Cheng VC, Hung IF, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax 2004; 59(3): 252-6.
66. Spanakis N, Tsiodras S, Haagmans BL, et al. Virological and serological analysis of a recent Middle East respiratory syndrome coronavirus infection case on a triple combination antiviral regimen. Int J Antimicrob Agents 2014; 44(6): 528-32.
67. Kim UJ, Won EJ, Kee SJ, Jung SI, Jang HC. Combination therapy with lopinavir/ritonavir, ribavirin and interferon-alpha for Middle East respiratory syndrome. Antivir Ther 2016; 21(5): 455-9.
68. Hung IF, Lung KC, Tso EY, et al. Triple combination of interferon beta-1b, lopinavir-ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet 2020; 395(10238): 1695-704.
69. Labhardt ND, Smit M, Petignat I, et al. Post-exposure Lopinavir-Ritonavir Prophylaxis versus Surveillance for Individuals Exposed to SARS-CoV-2: The COPEP Pragmatic Open-Label, Cluster Randomized Trial. EClinicalMedicine 2021; 42: 101188.
70. Reis G, Moreira Silva E, Medeiros Silva DC, et al. Effect of Early Treatment With Hydroxychloroquine or Lopinavir and Ritonavir on Risk of Hospitalization Among Patients With COVID-19: The TOGETHER Randomized Clinical Trial. JAMA Netw Open 2021; 4(4): e216468.
71. Cao B, Wang Y, Wen D, et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N Engl J Med 2020; 382(19): 1787-99.
72. RECOVERY Collaborative Group, Horby PW, Mafham M, et al. Lopinavir–ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. The Lancet 2020; 396(10259): 1345-52.
73. World Health Organization. Clinical management of severe acute respiratory infection (SARI) when COVID-19 disease is suspected. Available at: https://apps.who.int/iris/bitstream/handle/10665/331446/WHO-2019-nCoV-clinical-2020.4-eng.pdf. Accessed 24 June 2020.
74. Arabi YM, Mandourah Y, Al-Hameed F, et al. Corticosteroid Therapy for Critically Ill Patients with Middle East Respiratory Syndrome. Am J Respir Crit Care Med 2018; 197(6): 757-67.
75. Lee N, Allen Chan KC, Hui DS, et al. Effects of early corticosteroid treatment on plasma SARS-associated Coronavirus RNA concentrations in adult patients. J Clin Virol 2004; 31(4): 304-9.
76. Xiao JZ, Ma L, Gao J, et al. [Glucocorticoid-induced diabetes in severe acute respiratory syndrome: the impact of high dosage and duration of methylprednisolone therapy]. Zhonghua Nei Ke Za Zhi 2004; 43(3): 179-82.
77. Laurent A, Bonnet M, Capellier G, Aslanian P, Hebert P. Emotional Impact of End-of-Life Decisions on Professional Relationships in the ICU: An Obstacle to Collegiality? Crit Care Med 2017; 45(12): 2023-30.
78. Villar J, Ferrando C, Martinez D, et al. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med 2020; 8(3): 267-76.
79. WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group, Sterne JAC, Murthy S, et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020; 324(13): 1330-41.
80. Horby P, Lim WS, Emberson J, et al. Effect of Dexamethasone in Hospitalized Patients with COVID-19 – Preliminary Report. medRxiv 2020: Available at: https://doi.org/10.1101/2020.06.22.20137273 [Preprint 22 June 2020].
81. Tomazini BM, Maia IS, Cavalcanti AB, et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA 2020.
82. Dequin PF, Heming N, Meziani F, et al. Effect of Hydrocortisone on 21-Day Mortality or Respiratory Support Among Critically Ill Patients With COVID-19: A Randomized Clinical Trial. JAMA 2020.
83. Writing Committee for the REMAP-CAP Investigators, Angus DC, Derde L, et al. Effect of Hydrocortisone on Mortality and Organ Support in Patients With Severe COVID-19: The REMAP-CAP COVID-19 Corticosteroid Domain Randomized Clinical Trial. JAMA 2020.
84. Petersen MW, Meyhoff TS, Helleberg M, et al. Low-dose hydrocortisone in patients with COVID-19 and severe hypoxia (COVID STEROID) trial-Protocol and statistical analysis plan. Acta Anaesthesiol Scand 2020.
85. Jeronimo CMP, Farias MEL, Val FFA, et al. Methylprednisolone as Adjunctive Therapy for Patients Hospitalized With COVID-19 (Metcovid): A Randomised, Double-Blind, Phase IIb, Placebo-Controlled Trial. Clin Infect Dis 2020.
86. Corral-Gudino L, Bahamonde A, Arnaiz delas Revillas F, et al. GLUCOCOVID: A controlled trial of methylprednisolone in adults hospitalized with COVID-19 pneumonia. medRxiv 2020: Available at: https://doi.org/10.1101/2020.06.17.20133579 [Preprint 18 June 2020].
87. Salton F, Confalonieri P, Santus P, et al. Prolonged low-dose methylprednisolone in patients with severe COVID-19 pneumonia. medRxiv 2020: Available at: https://doi.org/10.1101/2020.06.17.20134031 [Preprint 25 June 2020].
88. Wang Y, Jiang W, He Q, et al. Early, low-dose and short-term application of corticosteroid treatment in patients with severe COVID-19 pneumonia: single-center experience from Wuhan, China. medRxiv 2020: Available at: https://doi.org/10.1101/2020.03.06.20032342 [Preprint 12 March 2020].
89. Wu C, Chen X, Cai Y, et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med 2020; 180(7): 934-43.
90. Fernandez-Cruz A, Ruiz-Antoran B, Munoz-Gomez A, et al. Impact of Glucocorticoid Treatment in SARS-CoV-2 Infection Mortality: A retrospective controlled cohort study. 2020: Available at: https://doi.org/10.1101/2020.05.22.20110544 [Preprint 26 May 2020].
91. Lu X, Chen T, Wang Y, et al. Adjuvant corticosteroid therapy for critically ill patients with COVID-19. medRxiv 2020: Available at: https://doi.org/10.1101/2020.04.07.20056390 [Preprint 11 April 2020].
92. Yuan M, Xu X, Xia D, et al. Effects of Corticosteroid Treatment for Non-Severe COVID-19 Pneumonia: A Propensity Score-Based Analysis. Shock 2020; 54(5): 638-43.
93. Henzen C, Suter A, Lerch E, Urbinelli R, Schorno XH, Briner VA. Suppression and recovery of adrenal response after short-term, high-dose glucocorticoid treatment. Lancet 2000; 355(9203): 542-5.
94. Siemieniuk RA, Meade MO, Alonso-Coello P, et al. Corticosteroid Therapy for Patients Hospitalized With Community-Acquired Pneumonia: A Systematic Review and Meta-analysis. Ann Intern Med 2015; 163(7): 519-28.
95. RECOVERY Collaborative Group, Horby P, Lim WS, et al. Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med 2021; 384(8): 693-704.
96. Matsuyama S, Kawase M, Nao N, et al. The Inhaled Steroid Ciclesonide Blocks SARS-CoV-2 RNA Replication by Targeting the Viral Replication-Transcription Complex in Cultured Cells. J Virol 2020; 95(1).
97. Peters MC, Sajuthi S, Deford P, et al. COVID-19-related Genes in Sputum Cells in Asthma. Relationship to Demographic Features and Corticosteroids. Am J Respir Crit Care Med 2020; 202(1): 83-90.
98. Yu LM, Bafadhel M, Dorward J, et al. Inhaled budesonide for COVID-19 in people at high risk of complications in the community in the UK (PRINCIPLE): a randomised, controlled, open-label, adaptive platform trial. Lancet 2021; 398(10303): 843-55.
99. Clemency BM, Varughese R, Gonzalez-Rojas Y, et al. Efficacy of Inhaled Ciclesonide for Outpatient Treatment of Adolescents and Adults With Symptomatic COVID-19: A Randomized Clinical Trial. JAMA Intern Med 2022; 182(1): 42-9.
100. Ezer N, Belga S, Daneman N, et al. Inhaled and intranasal ciclesonide for the treatment of covid-19 in adult outpatients: CONTAIN phase II randomised controlled trial. BMJ 2021; 375: e068060.
101. Song JY, Yoon JG, Seo YB, et al. Ciclesonide Inhaler Treatment for Mild-to-Moderate COVID-19: A Randomized, Open-Label, Phase 2 Trial. J Clin Med 2021; 10(16): 3545.
102. Ramakrishnan S, Nicolau DV, Jr., Langford B, et al. Inhaled budesonide in the treatment of early COVID-19 (STOIC): a phase 2, open-label, randomised controlled trial. Lancet Respir Med 2021; 9(7): 763-72.
103. Accelerating Covid-19 Therapeutic I, Vaccines -6 Study G, Naggie S. Inhaled Fluticasone for Outpatient Treatment of Covid-19: A Decentralized, Placebo-controlled, Randomized, Platform Clinical Trial. medRxiv 2022.
104. Agusti A, De Stefano G, Levi A, et al. Add-on inhaled budesonide in the treatment of hospitalised patients with COVID-19: a randomised clinical trial. Eur Respir J 2022; 59(3).
105. Duvignaud A, Lhomme E, Onaisi R, et al. Inhaled ciclesonide for outpatient treatment of COVID-19 in adults at risk of adverse outcomes: a randomised controlled trial (COVERAGE). Clin Microbiol Infect 2022; 28(7): 1010-6.
106. Boyd SD, Hadigan C, McManus M, et al. Influence of low-dose ritonavir with and without darunavir on the pharmacokinetics and pharmacodynamics of inhaled beclomethasone. J Acquir Immune Defic Syndr 2013; 63(3): 355-61.
107. Chen G, Wu D, Guo W, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 2020; 130(5): 2620-9.
108. Kalikshtein DB, Levantovskaia OM, Vyshenepol'skii I, Ol'shanskii A. [Coagulation and anticoagulation systems of the blood in allergic diseases]. Sov Med 1988; (9): 104-6.
109. REMAP-CAP Investigators, Gordon AC, Mouncey PR, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19 – Preliminary report. medRxiv 2021: Available at: https://doi.org/10.1101/2021.01.07.21249390 [Preprint 9 January 2021].
110. Hermine O, Mariette X, Tharaux PL, et al. Effect of Tocilizumab vs Usual Care in Adults Hospitalized With COVID-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern Med 2020; 181(1): 32-40.
111. Horby PW, Pessoa-Amorim G, Peto L, et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): preliminary results of a randomised, controlled, open-label, platform trial. Lancet 2021; 397(10285): 1637-45.
112. Rosas I, Bräu N, Waters M, et al. Tocilizumab in hospitalized patients with COVID-19 pneumonia. medRxiv 2020: Available at: https://doi.org/10.1101/2020.08.27.20183442 [Preprint 12 September 2020].
113. Salama C, Han J, Yau L, et al. Tocilizumab in Patients Hospitalized with Covid-19 Pneumonia. N Engl J Med 2021; 384(1): 20-30.
114. Salvarani C, Dolci G, Massari M, et al. Effect of Tocilizumab vs Standard Care on Clinical Worsening in Patients Hospitalized With COVID-19 Pneumonia: A Randomized Clinical Trial. JAMA Intern Med 2020; 181(1): 24-31.
115. Stone JH, Frigault MJ, Serling-Boyd NJ, et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N Engl J Med 2020; 383: 2333-44.
116. Veiga VC, Prats J, Farias DLC, et al. Effect of tocilizumab on clinical outcomes at 15 days in patients with severe or critical coronavirus disease 2019: randomised controlled trial. BMJ 2021; 372: n84.
117. Lescure FX, Honda H, Fowler RA, et al. Sarilumab in patients admitted to hospital with severe or critical COVID-19: a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med 2021; 9(5): 522-32.
118. Sivapalasingam S, Lederer D, Bhore R, et al. A Randomized Placebo-Controlled Trial of Sarilumab in Hospitalized Patients with Covid-19. medRxiv 2021: Available at: https://doi.org/10.1101/2021.05.13.21256973 [Preprint 14 May 2021].
119. Godolphin PJ, Fisher DJ, Berry LR, et al. Association between tocilizumab, sarilumab and all-cause mortality at 28 days in hospitalized patients with COVID-19: A network meta-analysis. medRxiv 2021: Available at: https://doi.org/10.1101/2021.08.26.21262523 [Preprint 28 August 2021].
120. Rojo M, Cano-Valderrama O, Picazo S, et al. Gastrointestinal Perforation After Treatment With Tocilizumab : An Unexpected Consequence of COVID-19 Pandemic. Am Surg 2020; 86(6): 565-6.
121. Gonzalvez Guardiola P, Diez Ares JA, Peris Tomas N, Sebastian Tomas JC, Navarro Martinez S. Intestinal perforation in patient with COVID-19 infection treated with tocilizumab and corticosteroids. Report of a clinical case. Cir Esp 2020.
122. Ranchal P, Yates E, Gupta R, Aronow WS. Tocilizumab-Associated Bowel Perforation in SARS-CoV-2 Infection. Am J Ther 2020.
123. Bruce-Hickman D, Sajeed SM, Pang YH, Seow CS, Chen W, Gulati Kansal M. Bowel ulceration following tocilizumab administration in a COVID-19 patient. BMJ Open Gastroenterol 2020; 7(1).
124. Guaraldi G, Meschiari M, Cozzi-Lepri A, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol 2020; 2(8): e474-e84.
125. Somers EC, Eschenauer GA, Troost JP, et al. Tocilizumab for treatment of mechanically ventilated patients with COVID-19. Clin Infect Dis 2020.
126. Li L, Zhang W, Hu Y, et al. Effect of Convalescent Plasma Therapy on Time to Clinical Improvement in Patients With Severe and Life-threatening COVID-19: A Randomized Clinical Trial. JAMA 2020; 324(5): 460-70.
127. Gharbharan A, Jordans CC, Geurts van Kessel C, et al. Effects of potent neutralizing antibodies from convalescent plasma in patients hospitalized for severe SARS-CoV-2 infection. Nat Commun 2021; 12(3189).
128. AlQahtani M, Abdulrahman A, AlMadani A, et al. Randomized controlled trial of convalescent plasma therapy against standard therapy in patients with severe COVID-19 disease. Sci Rep 2021; 11: 9927.
129. Avendaño-Solà C, Ramos-Martinez A, Munez-Rubio E, et al. A multicenter randomized open-label clinical trial for convalescent plasma in patients hospitalized with COVID-19 pneumonia. J Clin Invest 2021; 131(20).
130. Libster R, Marc GP, Wappner D, et al. Prevention of severe COVID-19 in the elderly by early high-titer plasma. medRxiv 2020: Available at: https://doi.org/10.1101/2020.11.20.20234013 [Preprint 21 November 2020].
131. Joyner MJ, Senefeld JW, Klassen SA, et al. Effect of convalescent plasma on mortality among hospitalized patients with COVID-19: initial three-month experience. medRxiv 2020: Available at: https://doi.org/10.1101/2020.08.12.20169359 [Preprint 12 August 2020].
132. Expanded Access to Convalescent Plasma for the Treatment of Patients Team, Joyner M. Convalescent Plasma COVID-19 (Coronavirus) Treatment. Available at: https://www.uscovidplasma.org/. Accessed 1 November 2021.
133. Bégin P, Callum J, Jamula E, et al. Convalescent plasma for hospitalized patients with COVID-19: an open-label, randomized controlled trial. Nat Med 2021: Available at: https://doi.org/10.1038/s41591-021-01488-2 [Epub ahead of print 9 September 2021].
134. U.S. Food and Drug Administration. Recommendations for Investigational COVID-19 Convalescent Plasma. Available at: https://www.fda.gov/vaccines-blood-biologics/investigational-new-drug-ind-or-device-exemption-ide-process-cber/recommendations-investigational-covid-19-convalescent-plasma. Accessed 28 August 2020.
135. U.S. Food and Drug Administration. FDA In Brief: FDA Updates Emergency Use Authorization for COVID-19 Convalescent Plasma to Reflect New Data. Available at: https://www.fda.gov/news-events/fda-brief/fda-brief-fda-updates-emergency-use-authorization-covid-19-convalescent-plasma-reflect-new-data. Accessed 30 March 2021.
136. Ray Y, Paul SR, Bandopadhyay P, et al. Clinical and immunological benefits of convalescent plasma therapy in severe COVID-19: insights from a single center open label randomised control trial. medRxiv 2020: Available at: https://doi.org/10.1101/2020.11.25.20237883 [Preprint 29 November 2020].
137. Simonovich VA, Burgos Pratx LD, Scibona P, et al. A Randomized Trial of Convalescent Plasma in Covid-19 Severe Pneumonia. N Engl J Med 2021; 384(7): 619-29.
138. Agarwal A, Mukherjee A, Kumar G, et al. Convalescent plasma in the management of moderate covid-19 in adults in India: open label phase II multicentre randomised controlled trial (PLACID Trial). BMJ 2020; 371: m4232.
139. O’Donnell MR, Grinsztejn B, Cummings MJ, et al. A randomized double-blind controlled trial of convalescent plasma in adults with severe COVID-19. J Clin Invest 2021; 131(13): e150646.
140. RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised controlled, open-label, platform trial. Lancet 2021; 397(10289): 2049-59.
141. Balcells ME, Rojas L, Le Corre N, et al. Early versus deferred anti-SARS-CoV-2 convalescent plasma in patients admitted for COVID-19: A randomized phase II clinical trial. PLoS Med 2021; 18(3): e1003415.
142. Joyner MJ, Bruno KA, Klassen SA, et al. Safety Update: COVID-19 Convalescent Plasma in 20,000 Hospitalized Patients. Mayo Clin Proc 2020; 95(9): 1888-97.
143. Korley FK, Durkalski-Mauldin V, Yeatts SD, et al. Early Convalescent Plasma for High-Risk Outpatients with Covid-19. N Engl J Med 2021; 385(21): 1951-60.
144. Libster R, Perez Marc G, Wappner D, et al. Early High-Titer Plasma Therapy to Prevent Severe Covid-19 in Older Adults. N Engl J Med 2021; 384(7): 610-8.
145. Sullivan DJ, Gebo KA, Shoham S, et al. Randomized Controlled Trial of Early Outpatient COVID-19 Treatment with High-Titer Convalescent Plasma. medRxiv 2021: Available at: https://doi.org/10.1101/2021.12.10.21267485 [Preprint 21 December 2021].
146. Denkinger CM, Janssen M, Schakel U, et al. Anti-SARS-CoV-2 antibody-containing plasma improves outcome in patients with hematologic or solid cancer and severe COVID-19: a randomized clinical trial. Nat Cancer 2023; 4(1): 96-107.
147. Lacombe K, Hueso T, Porcher R, et al. COVID-19 convalescent plasma to treat hospitalised COVID-19 patients with or without underlying immunodeficiency. medRxiv 2022: Available at: https://doi.org/10.1101/2022.08.09.22278329 [Preprint 27 October 2022].
148. de Candia P, Prattichizzo F, Garavelli S, et al. Effect of time and titer in convalescent plasma therapy for COVID-19. iScience 2021; 24(8): 102898.
149. U.S. Food and Drug Administration. FDA Issues Emergency Use Authorization for Convalescent Plasma as Potential Promising COVID–19 Treatment, Another Achievement in Administration’s Fight Against Pandemic. Available at: https://www.fda.gov/news-events/press-announcements/fda-issues-emergency-use-authorization-convalescent-plasma-potential-promising-covid-19-treatment. Accessed 4 November 2021.
150. Joyner MJ, Carter RE, Senefeld JW, et al. Convalescent Plasma Antibody Levels and the Risk of Death from Covid-19. N Engl J Med 2021; 384(11): 1015-27.
151. Salazar E, Christensen PA, Graviss EA, et al. Significantly Decreased Mortality in a Large Cohort of Coronavirus Disease 2019 (COVID-19) Patients Transfused Early with Convalescent Plasma Containing High-Titer Anti-Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Spike Protein IgG. Am J Pathol 2021; 191(1): 90-107.
152. Arnold Egloff SA, Junglen A, Restivo JS, et al. Convalescent plasma associates with reduced mortality and improved clinical trajectory in patients hospitalized with COVID-19. J Clin Invest 2021; 131(20).
153. Thompson MA, Henderson JP, Shah PK, et al. Association of Convalescent Plasma Therapy With Survival in Patients With Hematologic Cancers and COVID-19. JAMA Oncol 2021; 7(8): 1167-75.
154. Lo MK, Jordan R, Arvey A, et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci Rep 2017; 7: 43395.
155. Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 2017; 9(396).
156. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016; 531(7594): 381-5.
157. Williamson BN, Feldmann F, Schwarz B, et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020; 585(7824): 273-6.
158. Gottlieb RL, Vaca CE, Paredes R, et al. Early Remdesivir to Prevent Progression to Severe Covid-19 in Outpatients. N Engl J Med 2021; 386(4): 305-15.
159. Beigel JH, Tomashek KM, Dodd LE, et al. Remdesivir for the Treatment of Covid-19 - Final Report. N Engl J Med 2020; 383(19): 1813-26.
160. Wang Y, Zhang D, Du G, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020; 395(10236): 1569-78.
161. Goldman JD, Lye DCB, Hui DS, et al. Remdesivir for 5 or 10 Days in Patients with Severe Covid-19. N Engl J Med 2020; 383: 1827-37.
162. Goldman DL, Aldrich ML, Hagmann SHF, et al. Compassionate Use of Remdesivir in Children With Severe COVID-19. Pediatrics 2021; 147(5).
163. Gilead Sciences, Inc. Study to Evaluate the Safety, Tolerability, Pharmacokinetics, and Efficacy of Remdesivir (GS-5734™) in Participants From Birth to < 18 Years of Age With Coronavirus Disease 2019 (COVID-19) (CARAVAN). Available at: https://www.clinicaltrials.gov/ct2/show/NCT04431453. Accessed 18 November 2020.
164. Borrell B. New York clinical trial quietly tests heartburn remedy against coronavirus. Available at: https://www.sciencemag.org/news/2020/04/new-york-clinical-trial-quietly-tests-heartburn-remedy-against-coronavirus.
165. Freedberg DE, Conigliaro J, Wang TC, et al. Famotidine use is associated with improved clinical outcomes in hospitalized COVID-19 patients: A propensity score matched retrospective cohort study. Gastroenterology 2020; 159(3): 1129-31.
166. Brennan CM, Nadella S, Zhao X, et al. Oral famotidine versus placebo in non-hospitalised patients with COVID-19: a randomised, double-blind, data-intense, phase 2 clinical trial. Gut 2022; 71(5): 879-88.
167. Pahwani S, Kumar M, Aperna F, et al. Efficacy of Oral Famotidine in Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2. Cureus 2022; 14(2): e22404.
168. Jorgensen SCJ, Tse CLY, Burry L, Dresser LD. Baricitinib: A Review of Pharmacology, Safety, and Emerging Clinical Experience in COVID-19. Pharmacotherapy 2020; 40(8): 843-56.
169. Bekerman E, Neveu G, Shulla A, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J Clin Invest 2017; 127(4): 1338-52.
170. Neveu G, Ziv-Av A, Barouch-Bentov R, Berkerman E, Mulholland J, Einav S. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol 2015; 89(8): 4387-404.
171. Richardson P, Griffin I, Tucker C, et al. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020; 395(10223): e30-e1.
172. Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J Infect 2020; 81(2): 318-56.
173. Titanji BK, Farley MM, Mehta A, et al. Use of Baricitinib in Patients with Moderate and Severe COVID-19. Clin Infect Dis 2020.
174. Rodriguez-Garcia JL, Sanchez-Nievas G, Arevalo-Serrano J, Garcia-Gomez C, Jimenez-Vizuete JM, Martinez-Alfaro E. Baricitinib improves respiratory function in patients treated with corticosteroids for SARS-CoV-2 pneumonia: an observational cohort study. Rheumatology (Oxford) 2021; 60(1): 399-407.
175. Cantini F, Niccoli L, Nannini C, et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J Infect 2020; 81(4): 647-79.
176. Marconi VC, Ramanan AV, de Bono S, et al. Baricitinib plus Standard of Care for Hospitalized Adults with COVID-19. medRxiv 2021: Available at: https://doi.org/10.1101/2021.04.30.21255934 [Preprint 3 May 2021].
177. OLUMIANT (baricitinib) tablet, for oral use (package insert). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf. Accessed 23 December 2020.
178. King B, Maari C, Lain E, et al. Extended Safety Analysis of Baricitinib 2 mg in Adult Patients with Atopic Dermatitis: An Integrated Analysis from Eight Randomized Clinical Trials. Am J Clin Dermatol 2021; 22(3): 395-405.
179. Winthrop KL, Harigai M, Genovese MC, et al. Infections in baricitinib clinical trials for patients with active rheumatoid arthritis. Ann Rheum Dis 2020; 79(10): 1290-7.
180. Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood 2020; 135(23): 2033-40.
181. Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N Engl J Med 2021; 384: 795-807.
182. Marconi VC, Ramanan AV, de Bono S, et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med 2021; 9(12): 1407-18.
183. RECOVERY Collaborative Group, Horby PW, Emberson JR, et al. Baricitinib in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial and updated meta-analysis. medRxiv 2022: Available at: https://doi.org/10.1101/2022.03.02.22271623 [Preprint 3 March 2022].
184. Ely EW, Ramanan AV, Kartman CE, et al. Efficacy and safety of baricitinib plus standard of care for the treatment of critically ill hospitalised adults with COVID-19 on invasive mechanical ventilation or extracorporeal membrane oxygenation: an exploratory, randomised, placebo-controlled trial. Lancet Respir Med 2022; 10(4): 327-36.
185. Guimaraes PO, Quirk D, Furtado RH, et al. Tofacitinib in Patients Hospitalized with Covid-19 Pneumonia. N Engl J Med 2021; 385(5): 406-15.
186. U.S. Food and Drug Administration. Safety trial finds risk of blood clots in the lungs and death with higher dose of tofacitinib (Xeljanz, Xeljanz XR) in rheumatoid arthritis patients; FDA to investigate. Available at: https://www.fda.gov/drugs/drug-safety-and-availability/safety-trial-finds-risk-blood-clots-lungs-and-death-higher-dose-tofacitinib-xeljanz-xeljanz-xr. Accessed 23 July 2021.
187. U.S. Food and Drug Administration. FDA approves Boxed Warning about increased risk of blood clots and death with higher dose of arthritis and ulcerative colitis medicine tofacitinib (Xeljanz, Xeljanz XR). Available at: https://www.fda.gov/drugs/drug-safety-and-availability/fda-approves-boxed-warning-about-increased-risk-blood-clots-and-death-higher-dose-arthritis-and. Accessed 23 July 2021.
188. U.S. Food and Drug Administration. Initial safety trial results find increased risk of serious heart-related problems and cancer with arthritis and ulcerative colitis medicine Xeljanz, Xeljanz XR (tofacitinib). Available at: https://www.fda.gov/drugs/drug-safety-and-availability/initial-safety-trial-results-find-increased-risk-serious-heart-related-problems-and-cancer-arthritis. Accessed 23 July 2021.
189. U.S. Food and Drug Administration. Xeljanz, Xeljanz XR (tofacitinib): Drug Safety Communication - Initial Safety Trial Results Find Increased Risk of Serious Heart-related Problems and Cancer with Arthritis and Ulcerative Colitis Medicine. Available at: https://www.fda.gov/safety/medical-product-safety-information/xeljanz-xeljanz-xr-tofacitinib-drug-safety-communication-initial-safety-trial-results-find-increased. Accessed 23 July 2021.
190. Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res 2020; 178: 104787.
191. Bray M, Rayner C, Noel F, Jans D, Wagstaff K. Ivermectin and COVID-19: A report in Antiviral Research, widespread interest, an FDA warning, two letters to the editor and the authors' responses. Antiviral Res 2020; 178: 104805.
192. Zhang X, Song Y, Ci X, et al. Ivermectin inhibits LPS-induced production of inflammatory cytokines and improves LPS-induced survival in mice. Inflamm Res 2008; 57(11): 524-9.
193. Yan S, Ci X, Chen N, et al. Anti-inflammatory effects of ivermectin in mouse model of allergic asthma. Inflamm Res 2011; 60(6): 589-96.
194. Ahmed S, Karim MM, Ross AG, et al. A five-day course of ivermectin for the treatment of COVID-19 may reduce the duration of illness. Int J Infect Dis 2020; 103: 214-6.
195. Chachar AZK, Khan KA, Asif M, Tanveer K, Khaqan A, Basri R. Effectiveness of Ivermectin in SARS-CoV-2/COVID-19 Patients. Int J Sci 2020; 9(09): 31-5.
196. Chaccour C, Casellas A, Blanco-Di Matteo A, et al. The effect of early treatment with ivermectin on viral load, symptoms and humoral response in patients with non-severe COVID-19: A pilot, double-blind, placebo-controlled, randomized clinical trial. EClinicalMedicine 2021; 32: 100720.
197. Hashim HA, Maulood MF, Rasheed AM, Fatak DF, Kabah KK, Abdulamir AS. Controlled randomized clinical trial on using Ivermectin with Doxycycline for treating COVID-19 patients in Baghdad, Iraq. medRxiv 2020: Available at: https://doi.org/10.1101/2020.10.26.20219345 [Preprint 27 October 2020].
198. Podder CS, Chowdhury N, Sina MI, Ul Haque WMM. Outcome of ivermectin treated mild to moderate COVID-19 cases: a single-centre, open-label, randomised controlled study. IMC J Med Sci 2020; 14(2): 11-8.
199. Bukhari SKHS, Asghar A, Perveen N, et al. Efficacy of Ivermectin in COVID-19 Patients with Mild to Moderate Disease. medRxiv 2021: Available at: https://doi.org/10.1101/2021.02.02.21250840 [Preprint 5 February 2021].
200. Ravikirti, Roy R, Pattadar C, et al. Ivermectin as a potential treatment for mild to moderate COVID-19–A double blind randomized placebo-controlled trial. medRxiv 2021: Available at: https://doi.org/10.1101/2021.01.05.21249310 [Preprint 9 January 2021].
201. Lopez-Medina E, Lopez P, Hurtado IC, et al. Effect of Ivermectin on Time to Resolution of Symptoms Among Adults With Mild COVID-19: A Randomized Clinical Trial. JAMA 2021; 325(14): 1426-35.
202. Mohan A, Tiwari P, Suri T, Mittal S, Patel AA, Jain A. Ivermectin in mild and moderate COVID-19 (RIVET-COV): a randomized, placebo-controlled trial. Research Square 2021: Available at: https://doi.org/10.21203/rs.3.rs-191648/v1 [Preprint 2 February 2021].
203. Beltran Gonzalez JL, Gonzalez Gamez M, Mendoza Enciso EA, et al. Efficacy and Safety of Ivermectin and Hydroxychloroquine in Patients with Severe COVID-19: A Randomized Controlled Trial. Infect Dis Rep 2022; 14(2): 160-8.
204. Krolewiecki A, Lifschitz A, Moragas M, et al. Antiviral effect of high-dose ivermectin in adults with COVID-19: A proof-of-concept randomized trial. EClinicalMedicine 2021; 37: 100959.
205. Abd-Elsalam S, Noor RA, Badawi R, et al. Clinical study evaluating the efficacy of ivermectin in COVID-19 treatment: A randomized controlled study. J Med Virol 2021; 93(10): 5833-8.
206. Mahmud R, Rahman MM, Alam I, et al. Ivermectin in combination with doxycycline for treating COVID-19 symptoms: a randomized trial. J Int Med Res 2021; 49(5): 3000605211013550.
207. Vallejos J, Zoni R, Bangher M, et al. Ivermectin to prevent hospitalizations in patients with COVID-19 (IVERCOR-COVID19) a randomized, double-blind, placebo-controlled trial. BMC Infect Dis 2021; 21(1): 635.
208. Biber A, Harmelin G, Lev D, et al. The effect of ivermectin on the viral load and culture viability in early treatment of nonhospitalized patients with mild COVID-19 - a double-blind, randomized placebo-controlled trial. Int J Infect Dis 2022; 122: 733-40.
209. Reis G, Silva E, Silva DCM, et al. Effect of Early Treatment with Ivermectin among Patients with Covid-19. N Engl J Med 2022; 386(18): 1721-31.
210. Abbas KU, Muhammad S, Ding SF. The Effect of Ivermectin on Reducing Viral Symptoms in Patients with Mild COVID-19. Indian J Pharm Sci 2022; 84(1): Spl Issue 87-91.
211. Buonfrate D, Chesini F, Martini D, et al. High-dose ivermectin for early treatment of COVID-19 (COVER study): a randomised, double-blind, multicentre, phase II, dose-finding, proof-of-concept clinical trial. Int J Antimicrob Agents 2022; 59(2): 106516.
212. Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV)-6 Study Group, Naggie S. Ivermectin for Treatment of Mild-to-Moderate COVID-19 in the Outpatient Setting: A Decentralized, Placebo-controlled, Randomized, Platform Clinical Trial. medRxiv 2022: Available at: https://doi.org/10.1101/2022.06.10.22276252 [Preprint 12 June 2022].
213. Lim SCL, Hor CP, Tay KH, et al. Efficacy of Ivermectin Treatment on Disease Progression Among Adults With Mild to Moderate COVID-19 and Comorbidities: The I-TECH Randomized Clinical Trial. JAMA Intern Med 2022; 182(4): 426-35.
214. Manomaipiboon A, Pholtawornkulchai K, Pupipatpab S, et al. Efficacy and safety of ivermectin in the treatment of mild-to-moderate COVID-19 infection: A randomized, double blind, placebo, controlled trial. Research Square 2022: Available at: https://doi.org/10.21203/rs.3.rs-1290999/v1 [Preprint 2 February 2022].
215. Babalola OE, Bode CO, Ajayi AA, et al. Ivermectin shows clinical benefits in mild to moderate Covid19 disease: A randomised controlled double blind dose response study in Lagos. medRxiv 2021: Available at: https://doi.org/10.1101/2021.01.05.21249131 [Preprint 6 January 2021].
216. Niaee MS, Gheibi N, Namdar P, et al. Ivermectin as an adjunct treatment for hospitalized adult COVID-19 patients: A randomized multi-center clinical trial. 2020: Available at: https://doi.org/10.21203/rs.3.rs-109670/v1 [Preprint 24 November 2020].
217. Rezaie S. COVID-19 Update: Ivermectin. Available at: https://rebelem.com/covid-19-update-ivermectin/. Accessed 10 February 2021.
218. Shoumann WM, Hegazy AA, Nafae RM, et al. Use of Ivermectin as a Potential Chemoprophylaxis for COVID-19 in Egypt: A Randomized Clinical Trial. JCDR 2021; 15(2): OC27-OC32.
219. Peral de Bruno MdlA, Chala RE. Prophylaxis Covid-19 in Healthcare Agents by Intensive Treatment With Ivermectin and Iota-carrageenan (Ivercar-Tuc). Available at: https://clinicaltrials.gov/ct2/show/NCT04701710.
220. Raad H. In vivo use of ivermectin (IVR) for treatment for corona virus infected patients (COVID-19): a randomized controlled trial. Available at: http://www.chictr.org.cn/showprojen.aspx?proj=54707.
221. Manomaipiboon A, Pholtawornkulchai K, Poopipatpab S, et al. Efficacy and safety of ivermectin in the treatment of mild to moderate COVID-19 infection: a randomized, double-blind, placebo-controlled trial. Trials 2022; 23(1): 714.
222. Elshafie AH, Elsawah HK, Hammad M, et al. Ivermectin role in COVID-19 treatment (IRICT): single-center, adaptive, randomized, double-blind, placebo-controlled, clinical trial. Expert Rev Anti Infect Ther 2022; 20(10): 1341-50.
223. George B, Moorthy M, Kulkarni U, et al. Single Dose of Ivermectin is not Useful in Patients with Hematological Disorders and COVID-19 Illness: A Phase II B Open Labelled Randomized Controlled Trial. Indian J Hematol Blood Transfus 2022; 38(4): 615-22.
224. Rezai MS, Ahangarkani F, Hill A, et al. Non-effectiveness of Ivermectin on Inpatients and Outpatients With COVID-19; Results of Two Randomized, Double-Blinded, Placebo-Controlled Clinical Trials. Front Med (Lausanne) 2022; 9: 919708.
225. Angkasekwinai N, Rattanaumpawan P, Chayakulkeeree M, et al. Safety and Efficacy of Ivermectin for the Prevention and Treatment of COVID-19: A Double-Blinded Randomized Placebo-Controlled Study. Antibiotics (Basel) 2022; 11(6).
226. Mirahmadizadeh A, Semati A, Heiran A, et al. Efficacy of single-dose and double-dose ivermectin early treatment in preventing progression to hospitalization in mild COVID-19: A multi-arm, parallel-group randomized, double-blind, placebo-controlled trial. Respirology 2022; 27(9): 758-66.
227. Bramante CT, Huling JD, Tignanelli CJ, et al. Randomized Trial of Metformin, Ivermectin, and Fluvoxamine for Covid-19. N Engl J Med 2022; 387(7): 599-610.
228. Rosen DA, Seki SM, Fernandez-Castaneda A, et al. Modulation of the sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci Transl Med 2019; 11(478).
229. Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020; 583(7816): 459-68.
230. Ishima T, Fujita Y, Hashimoto K. Interaction of new antidepressants with sigma-1 receptor chaperones and their potentiation of neurite outgrowth in PC12 cells. Eur J Pharmacol 2014; 727: 167-73.
231. Geiser F, Conrad R, Imbierowicz K, et al. Coagulation activation and fibrinolysis impairment are reduced in patients with anxiety and depression when medicated with serotonergic antidepressants. Psychiatry Clin Neurosci 2011; 65(5): 518-25.
232. Duerschmied D, Suidan GL, Demers M, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013; 121(6): 1008-15.
233. Lenze EJ, Mattar C, Zorumski CF, et al. Fluvoxamine vs Placebo and Clinical Deterioration in Outpatients With Symptomatic COVID-19: A Randomized Clinical Trial. JAMA 2020; 324(22): 2292-300.
234. Reis G, dos Santos Moreira Silva EA, Medeiros Silva DC, et al. Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial. Lancet 2021; S2214-109X(21): 00448-4.
235. U.S. Food and Drug Administration. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Paxlovid™. Available at: https://www.fda.gov/media/155050/download. Accessed 22 December 2021.
236. Liu J, Pan X, Zhang S, et al. Efficacy and safety of Paxlovid in severe adult patients with SARS-Cov-2 infection: a multicenter randomized controlled study. Lancet Reg Health West Pac 2023; 33: 100694.
237. U.S. Food and Drug Administration. Important Paxlovid™ EUA Dispensing Information for Patients with Moderate Renal Impairment. Available at: https://www.fda.gov/media/155072/download. Accessed 3 November 2022.
238. Anderson AS, Caubel P, Rusnak JM, Investigators E-HT. Nirmatrelvir-Ritonavir and Viral Load Rebound in Covid-19. N Engl J Med 2022; 387(11): 1047-9.
239. Wong CKH, Lau KTK, Au ICH, et al. Viral burden rebound in hospitalised patients with COVID-19 receiving oral antivirals in Hong Kong: a population-wide retrospective cohort study. Lancet Infect Dis 2023.
240. Wang L, Berger NA, Davis PB, Kaelber DC, Volkow ND, Xu R. COVID-19 rebound after Paxlovid and Molnupiravir during January-June 2022. medRxiv 2022: Available at: https://doi.org/10.1101/2022.06.21.22276724 [Preprint 22 June 2022].
241. U.S. Food and Drug Administration. FDA Briefing Document. Available at: https://www.fda.gov/media/166197/download. Accessed 20 March 2023.
242. Malden DE, Hong V, Lewin BJ, et al. Hospitalization and Emergency Department Encounters for COVID-19 After Paxlovid Treatment - California, December 2021-May 2022. MMWR Morb Mortal Wkly Rep 2022; 71(25): 830-3.
243. Ranganath N, O'Horo JC, Challener DW, et al. Rebound Phenomenon After Nirmatrelvir/Ritonavir Treatment of Coronavirus Disease 2019 (COVID-19) in High-Risk Persons. Clin Infect Dis 2023; 76(3): e537-e9.
244. Painter WP, Holman W, Bush JA, et al. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity Against SARS-CoV-2. Antimicrob Agents Chemother 2021; 65(5): e02428-20.
245. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med 2021: Available at: https://doi.org/10.1056/nejmoa2116044 [Epub ahead of print 16 December 2021].
246. Butler CC, Hobbs FDR, Gbinigie OA, et al. Molnupiravir plus usual care versus usual care alone as early treatment for adults with COVID-19 at increased risk of adverse outcomes (PANORAMIC): an open-label, platform-adaptive randomised controlled trial. Lancet 2023; 401(10373): 281-93.
247. Khoo SH, FitzGerald R, Saunders G, et al. Molnupiravir versus placebo in unvaccinated and vaccinated patients with early SARS-CoV-2 infection in the UK (AGILE CST-2): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Infect Dis 2023; 23(2): 183-95.
248. Fischer WA, 2nd, Eron JJ, Jr., Holman W, et al. A Phase 2a clinical trial of Molnupiravir in patients with COVID-19 shows accelerated SARS-CoV-2 RNA clearance and elimination of infectious virus. Sci Transl Med 2021: eabl7430. Available at: https://doi.org/10.1126/scitranslmed.abl7430 [Epub ahead of print 23 December 2021].
249. Zou R, Peng L, Shu D, et al. Antiviral Efficacy and Safety of Molnupiravir Against Omicron Variant Infection: A Randomized Controlled Clinical Trial. Front Pharmacol 2022; 13: 939573.
250. U.S. Food and Drug Administration. Fact Sheet for Patients And Caregivers: Emergency Use Authorization (EUA) Of Molnupiravir For Coronavirus Disease 2019 (COVID-19). Available at: https://www.fda.gov/media/155055/download. Accessed 3 November 2022.
251. U.S. Food and Drug Administration. Frequently Asked Questions on the Emergency Use Authorization for Lagevrio (molnupiravir) for
Treatment of COVID-19 Available at: https://www.fda.gov/media/155056/download. Accessed 13 February 2023.
252. Molad Y. Update on colchicine and its mechanism of action. Curr Rheumatol Rep 2002; 4(3): 252-6.
253. Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout. Clin Ther 2014; 36(10): 1465-79.
254. Ding AH, Porteu F, Sanchez E, Nathan CF. Downregulation of tumor necrosis factor receptors on macrophages and endothelial cells by microtubule depolymerizing agents. J Exp Med 1990; 171(3): 715-27.
255. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440(7081): 237-41.
256. Cronstein BN, Molad Y, Reibman J, Balakhane E, Levin RI, Weissmann G. Colchicine alters the quantitative and qualitative display of selectins on endothelial cells and neutrophils. J Clin Invest 1995; 96(2): 994-1002.
257. Mareev VY, Orlova YA, Plisyk AG, et al. Proactive anti-inflammatory therapy with colchicine in the treatment of advanced stages of new coronavirus infection. The first results of the COLORIT study. Kardiologiia 2021; 61(2): 15-27.
258. Alsultan M, Obeid A, Alsamarrai O, et al. Efficacy of Colchicine and Budesonide in Improvement Outcomes of Patients with Coronavirus Infection 2019 in Damascus, Syria: A Randomized Control Trial. Interdiscip Perspect Infect Dis 2021; 2021: 2129006.
259. Lopes MI, Bonjorno LP, Giannini MC, et al. Beneficial effects of colchicine for moderate to severe COVID-19: a randomised, double-blinded, placebo-controlled clinical trial. RMD Open 2021; 7(1): e001455.
260. Diaz R, Orlandini A, Castellana N, et al. Effect of Colchicine vs Usual Care Alone on Intubation and 28-Day Mortality in Patients Hospitalized With COVID-19: A Randomized Clinical Trial. JAMA Netw Open 2021; 4(12): e2141328.
261. Deftereos SG, Giannopoulos G, Vrachatis DA, et al. Effect of Colchicine vs Standard Care on Cardiac and Inflammatory Biomarkers and Clinical Outcomes in Patients Hospitalized With Coronavirus Disease 2019: The GRECCO-19 Randomized Clinical Trial. JAMA Netw Open 2020; 3(6): e2013136.
262. RECOVERY Collaborative Group. Colchicine in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet Respir Med 2021; 9(12): 1419-26.
263. Gaitán-Duarte HG, Álvarez-Moreno C, Rincón-Rodríguez CJ, et al. Effectiveness of Rosuvastatin plus Colchicine, Emtricitabine/Tenofovir and a combination of them in Hospitalized Patients with SARS Covid-19. EClinicalMedicine 2022; 43: 101242.
264. Pascual-Figal DA, Roura-Piloto AE, Moral-Escudero E, et al. Colchicine in Recently Hospitalized Patients with COVID-19: A Randomized Controlled Trial (COL-COVID). Int J Gen Med 2021; 14: 5517-26.
265. Absalon-Aguilar A, Rull-Gabayet M, Perez-Fragoso A, et al. Colchicine Is Safe Though Ineffective in the Treatment of Severe COVID-19: a Randomized Clinical Trial (COLCHIVID). J Gen Intern Med 2022; 37(1): 4-14.
266. Gorial FI, Maulood MF, Abdulamir AS, Alnuaimi AS, Abdulrrazaq MK, Bonyan FA. Randomized controlled trial of colchicine add on to the standard therapy in moderate and severe corona virus Disease-19 infection. Ann Med Surg (Lond) 2022; 77: 103593.
267. Tardif J-C, Bouabdallaoui N, L’Allier PL, et al. Efficacy of colchicine in non-hospitalized patients with COVID-19. medRxiv 2021: Available at: https://doi.org/10.1101/2021.01.26.21250494 [Preprint 27 January 2021].
268. Dorward J, Yu L-M, Hayward G, et al. Colchicine for COVID-19 in adults in the community (PRINCIPLE): a randomised, controlled, adaptive platform trial. medRxiv 2021: Available at: https://doi.org/10.1101/2021.09.20.21263828 [Preprint 23 September 2021].
269. Swedish Orphan Biovitrum. Fact Sheet for Healthcare Providers: Emergency Use Authorization for Kineret. Available at: https://www.fda.gov/media/163075/download. Accessed 21 April 2023.
270. Elmekaty E, Maklad A, Abouelhassan R, et al. Efficacy of Anakinra in the Management of Patients with COVID-19 Infection: A Randomized Clinical Trial. medRxiv 2022: Available at: https://doi.org/10.1101/2022.07.04.22277207 [Preprint 6 July 2022].
271. Declercq J, Van Damme KFA, De Leeuw E, et al. Effect of anti-interleukin drugs in patients with COVID-19 and signs of cytokine release syndrome (COV-AID): a factorial, randomised, controlled trial. Lancet Respir Med 2021; 9(12): 1427-38.
272. Kyriazopoulou E, Poulakou G, Milionis H, et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med 2021; 27(10): 1752-60.
273. Kharazmi AB, Moradi O, Haghighi M, et al. A randomized controlled clinical trial on efficacy and safety of anakinra in patients with severe COVID-19. Immun Inflamm Dis 2022; 10(2): 201-8.
274. Corimuno-Collaborative group. Effect of anakinra versus usual care in adults in hospital with COVID-19 and mild-to-moderate pneumonia (CORIMUNO-ANA-1): a randomised controlled trial. Lancet Respir Med 2021; 9(3): 295-304.
275. Audemard-Verger A, Le Gouge A, Pestre V, et al. Efficacy and safety of anakinra in adults presenting deteriorating respiratory symptoms from COVID-19: A randomized controlled trial. PLoS One 2022; 17(8): e0269065.
276. Centers for Disease Control and Prevention. Underlying Medical Conditions Associated with Higher Risk for Severe COVID-19: Information for Healthcare Professionals. Available at: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-care/underlyingconditions.html. Accessed 20 June 2022.
277. WHO Rapid Evidence Appraisal for COVID-19 Therapies Working Group, Shankar-Hari M, Vale CL, et al. Association Between Administration of IL-6 Antagonists and Mortality Among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA 2021; 326(6): 499-518.
278. Ely EW, Ramanan AV, Kartman CE, et al. Baricitinib plus Standard of Care for Hospitalised Adults with COVID-19 on Invasive Mechanical Ventilation or Extracorporeal Membrane Oxygenation: Results of a Randomised, Placebo-Controlled Trial. medRxiv 2021: Available at: https://doi.org/10.1101/2021.10.11.21263897 [Preprint 12 October 2021].
279. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet 2020; 395(10223): 507-13.
280. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 2020; 395(10229): 1054-62.
281. Rojo JMC, Santos JMA, Núñez-Cortés JM, et al. Clinical characteristics of patients hospitalized with COVID-19 in Spain: results from the SEMI-COVID-19 Network. medRxiv 2020.
282. Argenziano MG, Bruce SL, Slater CL, et al. Characterization and clinical course of 1000 Patients with COVID-19 in New York: retrospective case series. medRxiv 2020.
283. Vaughn VM, Gandhi TN, Petty LA, et al. Empiric Antibacterial Therapy and Community-onset Bacterial Coinfection in Patients Hospitalized With Coronavirus Disease 2019 (COVID-19): A Multi-hospital Cohort Study. Clin Infect Dis 2021; 72(10): e533-e41.
284. Mason CY, Kanitkar T, Richardson CJ, et al. Exclusion of bacterial co-infection in COVID-19 using baseline inflammatory markers and their response to antibiotics. J Antimicrob Chemother 2021; 76(5): 1323-31.
285. Townsend L, Hughes G, Kerr C, et al. Bacterial pneumonia coinfection and antimicrobial therapy duration in SARS-CoV-2 (COVID-19) infection. JAC Antimicrob Resist 2020; 2(3): dlaa071.
286. Calderon-Parra J, Muino-Miguez A, Bendala-Estrada AD, et al. Inappropriate antibiotic use in the COVID-19 era: Factors associated with inappropriate prescribing and secondary complications. Analysis of the registry SEMI-COVID. PLoS One 2021; 16(5): e0251340.
287. Karaba SM, Jones G, Helsel T, et al. Prevalence of Co-infection at the Time of Hospital Admission in COVID-19 Patients, A Multicenter Study. Open Forum Infect Dis 2021; 8(1): ofaa578.
288. Langford BJ, So M, Raybardhan S, et al. Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin Microbiol Infect 2020; 26(12): 1622-9.
289. Adler H, Ball R, Fisher M, Mortimer K, Vardhan MS. Low rate of bacterial co-infection in patients with COVID-19. Lancet Microbe 2020; 1(2): e62.
290. Hughes S, Troise O, Donaldson H, Mughal N, Moore LSP. Bacterial and fungal coinfection among hospitalized patients with COVID-19: a retrospective cohort study in a UK secondary-care setting. Clin Microbiol Infect 2020; 26(10): 1395-9.
291. Garcia-Vidal C, Sanjuan G, Moreno-Garcia E, et al. Incidence of co-infections and superinfections in hospitalized patients with COVID-19: a retrospective cohort study. Clin Microbiol Infect 2021; 27(1): 83-8.
292. Baghdadi JD, Coffey KC, Adediran T, et al. Antibiotic Use and Bacterial Infection among Inpatients in the First Wave of COVID-19: a Retrospective Cohort Study of 64,691 Patients. Antimicrob Agents Chemother 2021; 65(11): e0134121.
293. Smith L, Karaba SM, Amoah J, et al. Hospital-acquired infections among adult patients admitted for coronavirus disease 2019 (COVID-19). Infect Control Hosp Epidemiol 2021: 1-4.
294. Kreitmann L, Monard C, Dauwalder O, Simon M, Argaud L. Early bacterial co-infection in ARDS related to COVID-19. Intensive Care Med 2020; 46(9): 1787-9.
295. Dudoignon E, Camelena F, Deniau B, et al. Bacterial Pneumonia in COVID-19 Critically Ill Patients: A Case Series. Clin Infect Dis 2021; 72(5): 905-6.
296. Fabre V, Karaba S, Amoah J, et al. The role of procalcitonin results in antibiotic decision-making in coronavirus disease 2019 (COVID-19). Infect Control Hosp Epidemiol 2021: 1-6.
297. Matsubara JA, Phillips DP. Intracortical connections and their physiological correlates in the primary auditory cortex (AI) of the cat. J Comp Neurol 1988; 268(1): 38-48.
298. Clancy CJ, Nguyen MH. COVID-19, superinfections and antimicrobial development: What can we expect? Clin Infect Dis 2020.
299. Centers for Disease Control and Prevention. Demographic Trends of COVID-19 cases and deaths in the US reported to CDC. Available at: https://covid.cdc.gov/covid-data-tracker/#demographics. Accessed 21 November 2021.
300. Stokes EK, Zambrano LD, Anderson KN, et al. Coronavirus Disease 2019 Case Surveillance - United States, January 22-May 30, 2020. MMWR Morb Mortal Wkly Rep 2020; 69(24): 759-65.
301. Kim L, Whitaker M, O'Halloran A, et al. Hospitalization Rates and Characteristics of Children Aged <18 Years Hospitalized with Laboratory-Confirmed COVID-19 - COVID-NET, 14 States, March 1-July 25, 2020. MMWR Morb Mortal Wkly Rep 2020; 69(32): 1081-8.
302. Han MS, Choi EH, Chang SH, et al. Clinical Characteristics and Viral RNA Detection in Children With Coronavirus Disease 2019 in the Republic of Korea. JAMA Pediatr 2021; 175(1): 73-80.
303. Chung E, Chow EJ, Wilcox NC, et al. Comparison of Symptoms and RNA Levels in Children and Adults With SARS-CoV-2 Infection in the Community Setting. JAMA Pediatr 2021; 175(10): e212025.
304. Delahoy MJ, Ujamaa D, Whitaker M, et al. Hospitalizations Associated with COVID-19 Among Children and Adolescents - COVID-NET, 14 States, March 1, 2020-August 14, 2021. MMWR Morb Mortal Wkly Rep 2021; 70(36): 1255-60.
305. Siegel DA, Reses HE, Cool AJ, et al. Trends in COVID-19 Cases, Emergency Department Visits, and Hospital Admissions Among Children and Adolescents Aged 0-17 Years - United States, August 2020-August 2021. MMWR Morb Mortal Wkly Rep 2021; 70(36): 1249-54.
306. Williams N, Radia T, Harman K, Agrawal P, Cook J, Gupta A. COVID-19 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in children and adolescents: a systematic review of critically unwell children and the association with underlying comorbidities. Eur J Pediatr 2021; 180(3): 689-97.
307. Hurst JH, Heston SM, Chambers HN, et al. Severe Acute Respiratory Syndrome Coronavirus 2 Infections Among Children in the Biospecimens from Respiratory Virus-Exposed Kids (BRAVE Kids) Study. Clin Infect Dis 2021; 73(9): e2875-e82.
308. Paul LA, Daneman N, Schwartz KL, et al. Association of Age and Pediatric Household Transmission of SARS-CoV-2 Infection. JAMA Pediatr 2021; 175(11): 1151-8.
309. Simon TD, Haaland W, Hawley K, Lambka K, Mangione-Smith R. Development and Validation of the Pediatric Medical Complexity Algorithm (PMCA) Version 3.0. Acad Pediatr 2018; 18(5): 577-80.
310. Leeb RT, Price S, Sliwa S, et al. COVID-19 Trends Among School-Aged Children - United States, March 1-September 19, 2020. MMWR Morb Mortal Wkly Rep 2020; 69(39): 1410-5.
311. Duarte-Salles T, Vizcaya D, Pistillo A, et al. Thirty-Day Outcomes of Children and Adolescents With COVID-19: An International Experience. Pediatrics 2021; 148(3).
312. Kompaniyets L, Agathis NT, Nelson JM, et al. Underlying Medical Conditions Associated With Severe COVID-19 Illness Among Children. JAMA Netw Open 2021; 4(6): e2111182.
313. Spinner CD, Gottlieb RL, Criner GJ, et al. Effect of Remdesivir vs Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020; 324(11): 1048-57.
314. Chiotos K, Hayes M, Kimberlin DW, et al. Multicenter Interim Guidance on Use of Antivirals for Children With Coronavirus Disease 2019/Severe Acute Respiratory Syndrome Coronavirus 2. J Pediatric Infect Dis Soc 2021; 10(1): 34-48.
315. Gotzinger F, Santiago-Garcia B, Noguera-Julian A, et al. COVID-19 in children and adolescents in Europe: a multinational, multicentre cohort study. Lancet Child Adolesc Health 2020; 4(9): 653-61.
316. Ahmed A, Rojo P, Agwu A, et al. Remdesivir Treatment for COVID-19 in Hospitalized Children: CARAVAN Interim Results. In: Conference on Retroviruses and Opportunistic Infections. Virtual, 2022.
317. REMAP-CAP Investigators, Gordon AC, Mouncey PR, et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with Covid-19. N Engl J Med 2021; 384(16): 1491-502.
318. Rosas IO, Brau N, Waters M, et al. Tocilizumab in Hospitalized Patients with Severe Covid-19 Pneumonia. N Engl J Med 2021; 384(16): 1503-16.
319. Bhumbra S, Malin S, Kirkpatrick L, et al. Clinical Features of Critical Coronavirus Disease 2019 in Children. Pediatr Crit Care Med 2020; 21(10): e948-e53.
320. Feldstein LR, Tenforde MW, Friedman KG, et al. Characteristics and Outcomes of US Children and Adolescents With Multisystem Inflammatory Syndrome in Children (MIS-C) Compared With Severe Acute COVID-19. JAMA 2021; 325(11): 1074-87.
321. Garcia-Salido A, de Carlos Vicente JC, Belda Hofheinz S, et al. Severe manifestations of SARS-CoV-2 in children and adolescents: from COVID-19 pneumonia to multisystem inflammatory syndrome: a multicentre study in pediatric intensive care units in Spain. Crit Care 2020; 24(1): 666.
322. U.S. Food and Drug Administration. Baricitinib EUA Letter of Authorization In: Eli Lilly and Company. Indianapolis, IN: Lilly Corporate Center, 2021.
323. Kim H, Brooks KM, Tang CC, et al. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin Pharmacol Ther 2018; 104(2): 364-73.
324. Eli Lilly and Company. A Study of Baricitinib (LY3009104) in Children With COVID-19 (COV-BARRIER-PEDS) (COV-BARRIER). Available at: https://clinicaltrials.gov/ct2/show/NCT05074420. Accessed 21 November 2021.
325. U.S. Food and Drug Administration. Highlights of Prescribing Information: XELJANZ® (tofacitinib) (package insert). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213082s000lbl.pdf. Accessed 11 August 2022.
326. Hammond J, Leister-Tebbe H, Gardner A, et al. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19. N Engl J Med 2022; 386(15): 1397-408.
327. Belhadjer Z, Meot M, Bajolle F, et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 2020; 142(5): 429-36.
328. Deza Leon MP, Redzepi A, McGrath E, et al. COVID-19-Associated Pediatric Multisystem Inflammatory Syndrome. J Pediatric Infect Dis Soc 2020; 9(3): 407-8.
329. Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet (London, England) 2020; 395(10237): 1607-8.
330. Verdoni L, Mazza A, Gervasoni A, et al. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. The Lancet 2020; 395(10239): 1771-8.
331. Blondiaux E, Parisot P, Redheuil A, et al. Cardiac MRI of Children with Multisystem Inflammatory Syndrome (MIS-C) Associated with COVID-19: Case Series. Radiology 2020: 202288.
332. Greene AG, Saleh M, Roseman E, Sinert R. Toxic shock-like syndrome and COVID-19: A case report of multisystem inflammatory syndrome in children (MIS-C). Am J Emerg Med 2020.
333. Pouletty M, Borocco C, Ouldali N, et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (Kawa-COVID-19): a multicentre cohort. Ann Rheum Dis 2020; 79(8): 999-1006.
334. Chiotos K, Bassiri H, Behrens EM, et al. Multisystem Inflammatory Syndrome in Children During the Coronavirus 2019 Pandemic: A Case Series. J Pediatric Infect Dis Soc 2020; 9(3): 393-8.
335. Cabrero-Hernandez M, Garcia-Salido A, Leoz-Gordillo I, et al. Severe SARS-CoV-2 Infection in Children With Suspected Acute Abdomen: A Case Series From a Tertiary Hospital in Spain. Pediatr Infect Dis J 2020; 39(8): e195-e8.
336. Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N Engl J Med 2020; 383(4): 334-46.
337. Dufort EM, Koumans EH, Chow EJ, et al. Multisystem Inflammatory Syndrome in Children in New York State. New England Journal of Medicine 2020; 383(4): 347-58.
338. Whittaker E, Bamford A, Kenny J, et al. Clinical Characteristics of 58 Children With a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA 2020; 324(3): 259-69.
339. Godfred-Cato S, Bryant B, Leung J, et al. COVID-19-Associated Multisystem Inflammatory Syndrome in Children - United States, March-July 2020. MMWR Morb Mortal Wkly Rep 2020; 69(32): 1074-80.
340. Payne AB, Gilani Z, Godfred-Cato S, et al. Incidence of Multisystem Inflammatory Syndrome in Children Among US Persons Infected With SARS-CoV-2. JAMA Netw Open 2021; 4(6): e2116420.
341. Stierman B, Abrams JY, Godfred-Cato SE, et al. Racial and Ethnic Disparities in Multisystem Inflammatory Syndrome in Children in the United States, March 2020 to February 2021. Pediatr Infect Dis J 2021; 40(11): e400-e6.
342. Belay ED, Abrams J, Oster ME, et al. Trends in Geographic and Temporal Distribution of US Children With Multisystem Inflammatory Syndrome During the COVID-19 Pandemic. JAMA Pediatr 2021; 175(8): 837-45.
343. Harwood R, Allin B, Jones CE, et al. A national consensus management pathway for paediatric inflammatory multisystem syndrome temporally associated with COVID-19 (PIMS-TS): results of a national Delphi process. Lancet Child Adolesc Health 2021; 5(2): 133-41.
344. Ouldali N, Toubiana J, Antona D, et al. Association of Intravenous Immunoglobulins Plus Methylprednisolone vs Immunoglobulins Alone With Course of Fever in Multisystem Inflammatory Syndrome in Children. JAMA 2021; 325(9): 855-64.
345. Kaushik S, Aydin SI, Derespina KR, et al. Multisystem Inflammatory Syndrome in Children Associated with Severe Acute Respiratory Syndrome Coronavirus 2 Infection (MIS-C): A Multi-institutional Study from New York City. J Pediatr 2020; 224: 24-9.
346. Capone CA, Subramony A, Sweberg T, et al. Characteristics, Cardiac Involvement, and Outcomes of Multisystem Inflammatory Syndrome of Childhood Associated with severe acute respiratory syndrome coronavirus 2 Infection. J Pediatr 2020; 224: 141-5.
347. Celikel E, Tekin ZE, Aydin F, et al. Role of Biological Agents in the Treatment of SARS-CoV-2-Associated Multisystem Inflammatory Syndrome in Children. J Clin Rheumatol 2022; 28(2): e381-e7.
348. Centers for Disease Control and Prevention. Health Department-Reported Cases of Multisystem Inflammatory Syndrome in Children (MIS-C) in the United States. Available at: https://covid.cdc.gov/covid-data-tracker/#mis-national-surveillance. Accessed 28 November 2021.